Keywords
Bacterial genomics, Bacterial functionome, Orthology, Horizontal gene transfer, clustering, semantic annotation
This article is included in the Bioinformatics gateway.
Bacterial genomics, Bacterial functionome, Orthology, Horizontal gene transfer, clustering, semantic annotation
We have amended the manuscript as suggested by the reviewer.
Specifically:
See the authors' detailed response to the review by David M. Kristensen
See the authors' detailed response to the review by Antonio Rosato
See the authors' detailed response to the review by Robert D. Finn
Comparative analysis of genome sequences has been pivotal to unravel mechanisms shaping bacterial evolution like gene duplication, loss and acquisition1,2, and helped in shedding light on pathogenesis and genotype-phenotype associations3,4.
Comparative analysis relies on the identification of sets of orthologous and paralogous genes and subsequent transfer of function to the encoding proteins. Technically orthologs are defined as best bi-directional hits (BBH) obtained via pairwise sequence comparison among multiple species and thus exploits sequence similarity for functional grouping. Sequence similarity-based (SB) methods present a number of shortcomings. First, a generalized minimal alignment length and similarity cut-off need to be arbitrarily selected for all, which may hamper proper functional grouping. Second, sequence and function might differ across evolutionary scales. Protein sequences change faster than protein structure and proteins with same function but with low sequence similarity have been identified5,6. SB methods may fail to group them hampering a functional comparison. This limitation becomes even more critical when comparing either phylogenetically distant genomes or gene sequences that were acquired with horizontal gene transfer events. Recent technological advancements are resulting in thousands of organisms and billions of proteins being sequenced7 which increases the need of methods able to perform comparisons at the larger scales.
To overcome these bottlenecks, protein domains have been suggested as an alternative for defining groups of functionally equivalent proteins8–10 and have been used to perform comparative analyses of Escherichia coli9, Pseudomonas10, Streptococcus11 and for protein functional annotation12,13. A protein domain architecture describes the arrangement of domains contained in a protein and is exemplified in Figure 1. As protein domains capture key structural and functional features, protein domain architectures may be considered to be better proxies to describe functional equivalence than a global sequence similarity14. The concept of using the domain architecture to precisely describe the extent of functional equivalence is exemplified in Figure 2. Moreover, once the probabilistic domain models have been defined, mining large sets of individual genome sequences for their occurrences is a considerably less demanding computational task than an exploration of all possible bi-directional hits between them15,16.
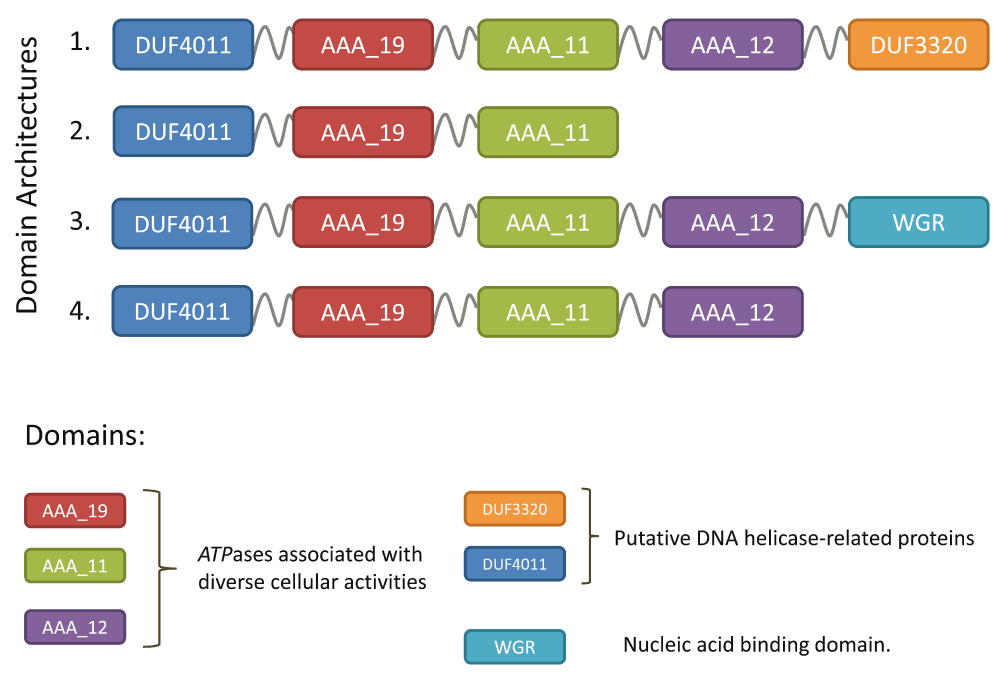
Although the proteins obviously share a common core, four distinct domain architectures involving six protein domains were observed in (1) Enterobacteriaceae, (2) H. pylori, (3) Pseudomonas and (4) Cyanobacteria.

Domains are probabilistic models of amino acids coordinates obtained by hidden Markov modeling (HMM) built from (structure based) multiple sequence alignments. Domain architectures are linear combinations of these domains representing the functional potential of a given protein sequence and constitute the input for DAB clustering. SB-orthology clusters inherit functional annotations via best bi-directional hits above a predefined sequence similarity cut-off score.
Domain architectures have been shown to be preserved at large phylogenetic distances both in prokaryotes and eukaryotes17,18. This lead to the use of protein domain architectures to classify and identify evolutionarily related proteins and to detect homologs even across evolutionarily distant species19–22. Structural information encoded in domain architectures has also been deployed to accelerate sequence search methods and to provide better homology detection. Examples are CDART23 which finds homologous proteins across significant evolutionary distances using domain profiles rather than direct sequence similarity, or DeltaBlast24 where a database of pre-constructed position-specific score matrix is queried before searching a protein-sequence database. Considering protein domain content, order, recurrence and position has been shown to increase the accuracy of protein function prediction25 and has led to the development of tools for protein functional annotation, such as UniProt-DAAC26 which uses domain architecture comparison and classification for the automatic functional annotation of large protein sets. The systematic assessment and use of domain architectures is enabled by databases containing protein domain information such as UniProt27, Pfam28, TIGRFAMs29, InterPro30, SMART31 and PROSITE32, that also provide graphical view of domain architectures.
Building on these observations we aim at exploring the potential of domain architecture-based (DAB) methods for large scale functional comparative analysis by comparing functionally equivalent sets of proteins, defined using domain architectures, with standard clusters of orthogonal proteins obtained with SB methods. We compared the SB and DAB approach by analysing i) the retrieved number of singletons (i.e. clusters containing only one protein) and ii) the characteristics of the inferred pan- and core-genome size considering a selection of bacterial genomes (both gram positive and negative) sampled at different taxonomic levels (species, genus, family, order and phylum). We show that the DAB approach provides a fast and efficient alternative to SB methods to identify groups of functionally equivalent/related proteins for comparative genome analysis and that the functional pan-genome is more closed in comparison to the sequence based pan-genome. DAB approaches can complement standardly applied sequence similarity methods and can pinpoint potential functional adaptations.
Bacterial species were chosen on the basis of the availability of fully sequenced genomes in the public domain: two species (Listeria monocytogenes and Helicobacter pylori), three genera (Streptococcus, Pseudomonas, Bacillus), one family (Enterobacteriaceae), one order (Corynebacteriales), and one phylum (Cyanobacteria) were selected. For each, 60 genome sequences were considered, except for L. monocytogenes for which only 26 complete genome sequences were available. Maximal diversity among genome sequences was ensured by sampling divergent species (when possible) at each taxonomic level. Genome sequences were retrieved from the European Nucleotide Archive database (www.ebi.ac.uk/ena). A full list of genomes analyzed is available in the Data availability section.
To avoid bias due to different algorithms used for the annotation of the original deposited genome sequences, all genomes were de novo re-annotated using the SAPP framework (1.0.0)10. In particular, the FASTA2RDF, GeneCaller (implementing Prodigal (2.6.2)33) and InterPro (implementing interproscan-5.17-56.0)34) modules were used to handle, re-annotate the genome sequences and store the results in the RDF data model. This resulted in 446 annotated genomes (7 × 60 genomes + 1 × 26 genomes). For each annotation step the provenance information (E-value cut off, score, originating tool or database) was stored together with annotation information in a graph database (RDF-model) and can be reproduced through the SAPP framework (http://semantics.systemsbiology.nl).
The positions (start and end on the protein sequence) of domains having Pfam28, TIGRFAMs29 and InterPro30 identifiers were extracted through SPARQL querying of the graph database and domain architectures were retrieved for each protein individually. InterPro aggregates protein domain signatures from different databases. Here no pruning for redundancies has been done. Identification of domains was done using the intrinsic InterPro cut-off that represents in each case the e-values and the scoring systems of the member databases30. The domain starting position was used to assess relative position in the case of overlapping domains; alphabetic ordering was used to order domains with the same starting position or when the distance between the starting position of overlapping domains was < 3 amino acids.
Labels indicating N-C terminal order of identified domains were assigned to each protein using the starting position of the domains: the same labels were assigned to proteins sharing the same domain architecture.
To make a direct comparison possible only protein sequences containing at least one protein domain signature were considered for analysis. BBH were obtained using Blastp (2.2.28+) with an E-value cutoff of 10−5 and -max_target_seqs of 105. OrthaGogue (1.0.3)35 combined with MCL (14-137)36 was used to identify protein clusters on the base of sequence similarity.
Domain architecture based clusters were built by clustering proteins with the same labels using bash terminal commands (sort, awk). The number of proteins sharing a given domain architecture in each genome was stored in a 446 × 21054 (genomes × domain architectures) matrix; from this a binarized presence-absence matrix was obtained and used solely for principal component analysis.
A Heaps’ law model was fit to the abundance matrices using 5 × 103 random genome ordering permutations and the micropan R package37.
SAPP, a Semantic Annotation Pipeline with Provenance which stores results in a graph database10, used for genome handling and annotation, is available at http://semantics.systemsbiology.nl. Matrix manipulations and multivariate analysis were performed using the R software (3.2.2).
SB and DAB approaches were compared by considering eight sets of genome sequences sampled at different taxonomic levels, from species to order, preserving phylogenetic diversity (see Table 1). Each set contained 60 genome sequences, except for Listeria monocytogenes for which only 26 complete genomes were publicly available. To facilitate the comparison between DAB and SB clusters only protein sequences that contained at least one domain were considered. On average, 85% of the protein sequences contain at least one domain from the InterPro database (see Table 1). Values range from 77±4% for Cyanobacteria to 91 ± 4% for Enterobacteriaceae (which include E. coli). Since the overall results were the same for gram negative and gram positive bacteria, we will show and comment only on results for the latter. Results obtained for gram negative bacteria are shown in the Data availability section.
DAB has been performed using HMM from Pfam (29.0) and InterPro (interproscan-5.17-56.0). Fraction refers to the fraction of proteins with at least one (InterPro or Pfam) protein domain. Core- and pan- indicate the sizes of the core- and pan- genomes (based on the sample) and singletons refers to the number of clusters with only one protein.
A standard BBH workflow was used to obtain SB protein clusters for the eight sets. We started by calculating the total number of clusters, corresponding to the pan-genome size, as shown in Table 1. Then we considered protein cluster persistence, that is the number of genomes where at least one member of the cluster is present, divided by the total number of genomes considered. Results are shown in Figure 3.
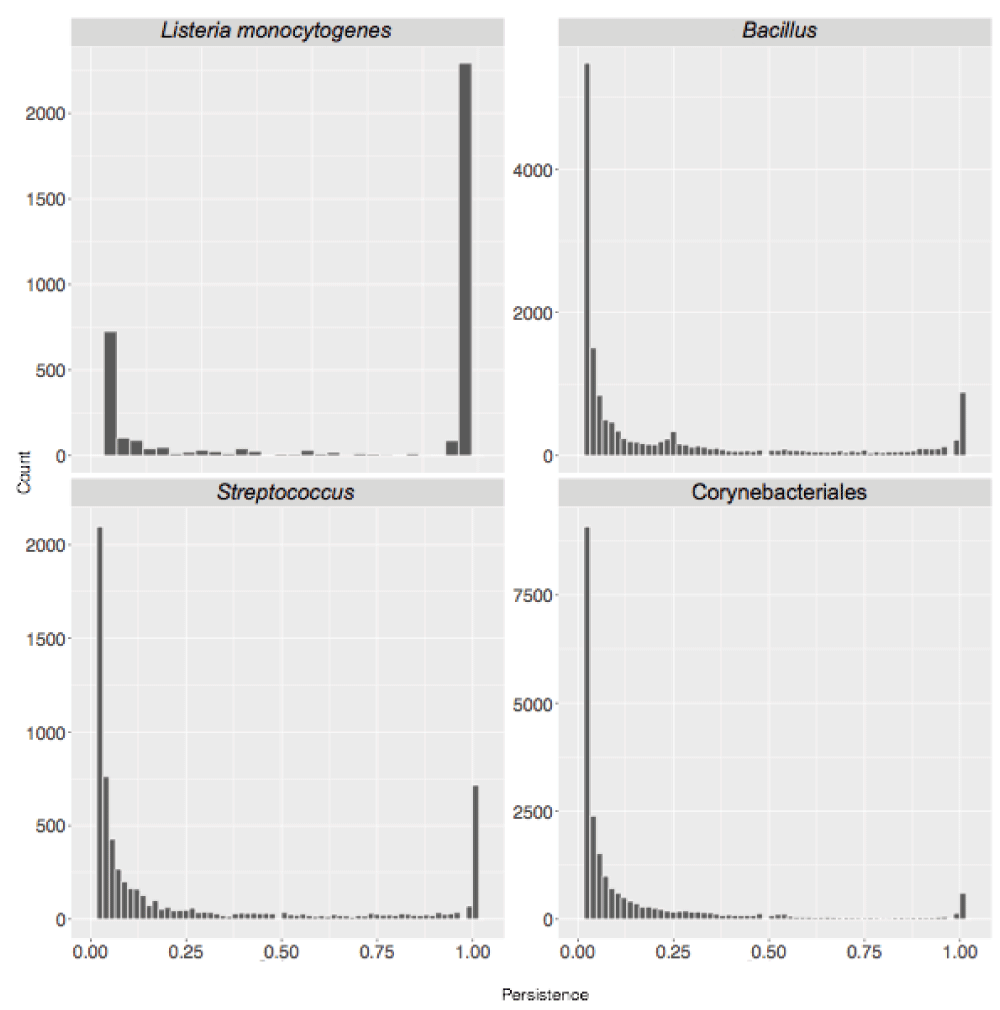
Cluster persistence is defined as the relative number of genomes with at least one protein assigned to the cluster. The frequency of SB clusters according to their persistence is shown.
The ratio between the size of the core-genome (clusters with persistence of 1, i.e. present in all genomes) and the number of singletons decreased with evolutionary distance (see Table 1). It ranged from 3.51 and 3.07 at species level (H. pylori and L. monocytogenes respectively) to 0.05 and 0.06 when considering members of the same order (Corynebacteriales) and phylum (Cyanobacteria) respectively. A similar pattern is observed when directly comparing the sizes of the pan- and core- genomes of the sampled genomes. Within the gram negative bacteria this ratio ranges from 0.69 for members of the same species (H. pylori) to 0.05 for members of the same phylum (Cyanobacteria) with intermediate values (0.12) for sequences from the same genus (Pseudomonas).
Domain architectures directly rely on the definition of protein domain models: those were retrieved from Pfam, InterPro and TIGRFAMs databases. However, TIGRFAMs results were not further considered because of a lower coverage. As shown in Table 1, as expected partly overlapping results were obtained when different domain databases were used. The number of singletons was larger when using InterPro rather than Pfam and for the latter we also observed larger core-genome size. These discrepancies can be due to the fact InterPro aggregates different resources (including Pfam and TIGRFAMs) and domain signatures arising from different databases are integrated with different identifiers in InterPro. In light of this we focused on results obtained using Pfam whose current release (30.0) contains hidden Markov models for over 16300 domain families. Size and persistence of groups of functionally equivalent proteins obtained using Pfam domains are presented in Figure 4.

The frequency of DAB clusters according to their persistence is shown.
Similar to what has been observed in the SB case we observed a decrease of the ratio between the size of the core genome and the number of singletons when higher taxonomic levels are considered. For organisms of the same species (H. pylori and L. monocytogenes) the ratio was 5.09 and 4.30, respectively, while for member of the same order (Corynebacteriales) and phylum (Cyanobacteria) it was 0.55 and 0.009 respectively. Similarly, also the ratio between the size of the core- and pan-genome decreases as higher taxonomic levels are considered, ranging from 0.54 for H. pylori to 0.04 for Cyanobacteria.
We compared the clusters obtained using both approaches and the proteins assigned to them. The number of one-to-one relationships (indicating a complete agreement) between SB and DAB clusters is indicated in Table 2 and ranges from 648 (for H. pylori) to 1680 (in Pseudomonas) corresponding to 50% and 25% of the pan-genome. This indicates that results of SB and DAB clustering tend to be more similar when working at closer phylogenetic distances. However, more complicated cases occur when proteins in a single SB cluster are assigned to various DAB clusters including singletons and vice versa. An overview of the possible mismatches between SB and DAB clusters is in Figure 5. The observed frequency of the different types of cluster mismatches are given in Figure 6. We observed that single domain architectures predominated the one-to-one clusters as shown in Table 3.

Mismatches of SB and DAB derived clusters (marked by S and D respectively) can occur in two directions. Panel A: possible cases of mismatch when counting the number of SB clusters the sequences in a DAB cluster are assigned to. 1d → 1s denotes that all sequences from the D cluster are assigned to the same S cluster. 1d → Ns denotes that sequences in a single D cluster are assigned to N distinct S clusters with N ≥ 1. Similarly, (panel B) 1s → Nd denotes that sequences in a single S cluster are assigned to N distinct D clusters with N ≥ 1.
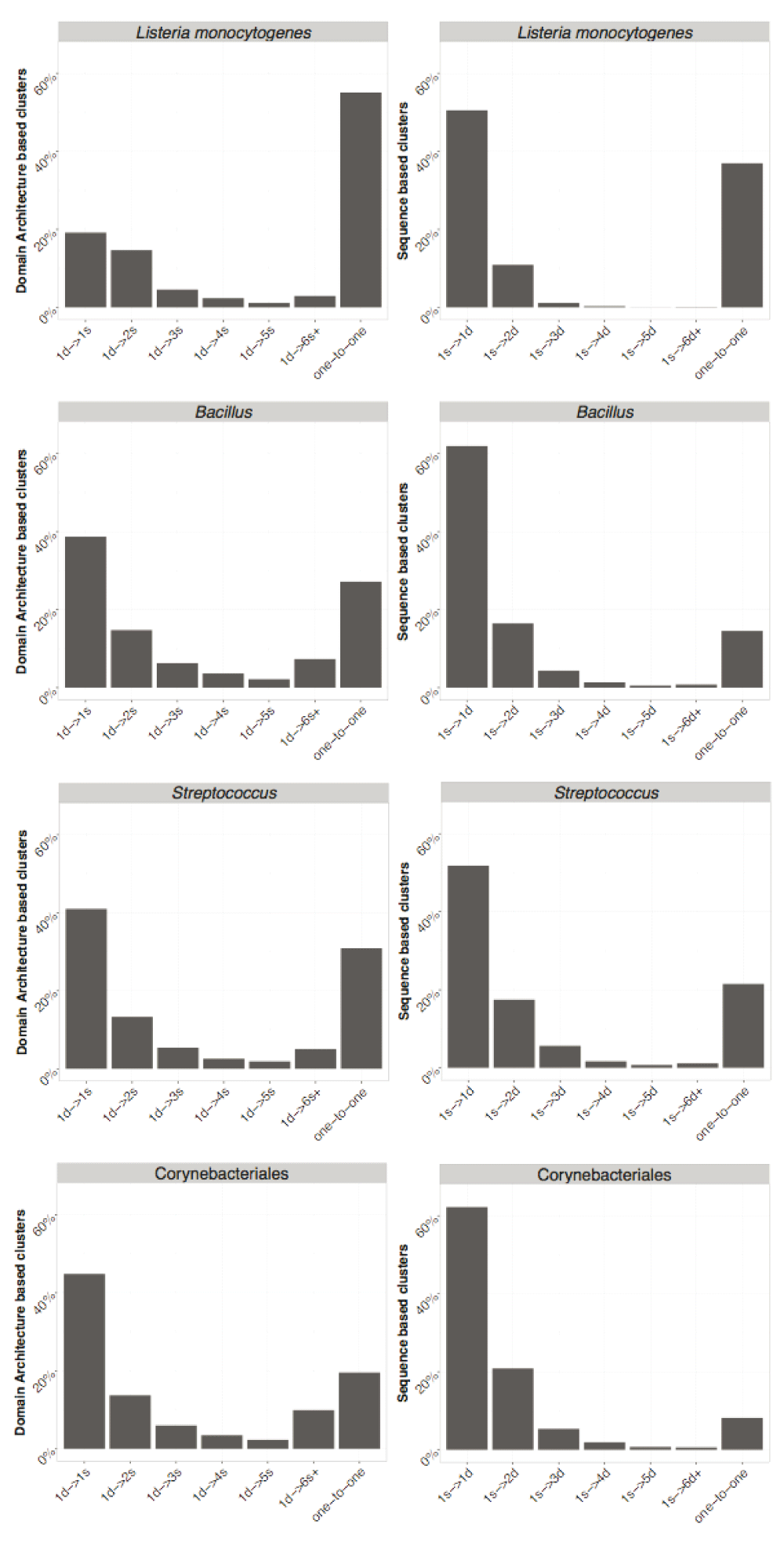
On the left DAB is used as a reference and each bar represents the relative frequency of one DAB cluster containing sequences assigned to {1, 2, ... , 5} and 6 or more SB clusters and one-to-one represents the relative frequency of identical cluster. Similarly, on the right SB is used as a reference. Axis labels follow notation in Figure 5.
| Group | Clusters |
|---|---|
| H. pylori | 648 |
| L. monocytogenes | 1085 |
| Bacillus | 1439 |
| Pseudomonas | 1680 |
| Streptococcus | 961 |
| Enterobacteriaceae | 1649 |
| Corynebacteriales | 1034 |
| Cyanobacteria | 1127 |
For L. monocytogenes we found 378 1d → 1s DAB cluster mismatches, (Figure 5, panel A, top case) meaning that in those cases sequences in a DAB cluster are a subset of the sequences in the corresponding SB cluster. This lower number of sequences in the DAB cluster could be due to, for instance an insertion or expansion of a domain, leading to SB clustered sequences with partly overlapping but distinct domain architectures as is depicted in Figure 1. Similarly, there are 399 1s → 1d clusters. Each of these cases represent a sequence cluster where all the sequences share the same domain architecture, but other sequences exist with the same architecture that have not been included in the cluster due to a too low similarity score. The low similarity between sequences with the same domain architecture could be due to a horizontal acquisition of the gene or to a fast protein evolution at the sequence level. Genes acquired from high phylogenetic distances could greatly vary in sequence while presenting the same domain architecture.
Proteins contained in a single DAB cluster but assigned to multiple SB clusters contain mostly ABC transporters-like (PF00005) or Major Facilitator Superfamily (MFS, PF07690) domains. This is not surprising considering that such generic functions are usually associated with a high sequence diversity. Conversely, ABC transporters are found in multiple DAB clusters. However, many of them are grouped into a single SB cluster with ATPase domain containing proteins (1s → Nd case).
We observed distinct architectures with one of two very similar domains, the GDSL-like Lipase/Acylhydrolase and the GDSL-like Lipase/Acylhydrolase family domain (PF00657 and PF13472 respectively) and those architectures were often seen clustered using a SB approach. However, architectures containing both domains were also identified, pointing to a degree of functional difference as a result of convergent or divergent evolution. Still, the corresponding sequences remain similar enough as to be indistinguishable when a SB approach is used. For SB clustering we also observed the case of identical protein sequences not clustered together, probably because of the tie breaking implementation when BBH are scored.
In all cases we found the size of both the pan- and the core-genome to be larger when a SB approach is used to identify gene clusters and SB approaches lead to a larger number of singletons than DAB ones. This indicates that DAB clusters are assigned to several SB clusters, many of them consisting of just one protein.
When going from species to phylum level, the ratio between the number of DAB and SB singletons changes from 0.48 and 0.41 (for H. pylori and L. monocytogenes respectively) to 0.19 and 0.40 when considering organisms of a higher taxonomic level (Corynebacteriales and Cyanobacteria respectively).
We investigated the predicted size of the pan-genome upon addition of new sequences. Heaps’ law regression can be used to estimate whether the pan-genome is open or closed38 through the fitting of the decay parameter α; α < 1 indicates openness of the pan-genome (indicating that possibly many clusters remain to be identified within the considered set of sequences), while α > 1 indicates a closed one; the α values are given in Table 4. In all cases the pan-genome is predicted to be open; however, α values obtained using DAB clusters (αDAB) are systematically closer to one than the αSB obtained with the standard sequence similarity approach.
The αDAB value retrieved for L. monocytogenes is strikingly low. Heaps law regression relies on the selected genomes providing a uniform sampling of selected taxon, here species. Analysis of the domain content of the selected genomes shows a divergent behaviour of strain LA111 (genome id GCA_000382925-1). This behaviour is clear in Figure 7, where GCA_000382925-1 appears as an outlier of the L. monocytogenes group. Removal of these outlier leads to αDAB = 1.04 and αSB = 0.64, which emphasizes the need for uniform sampling prior to Heaps regression analysis.
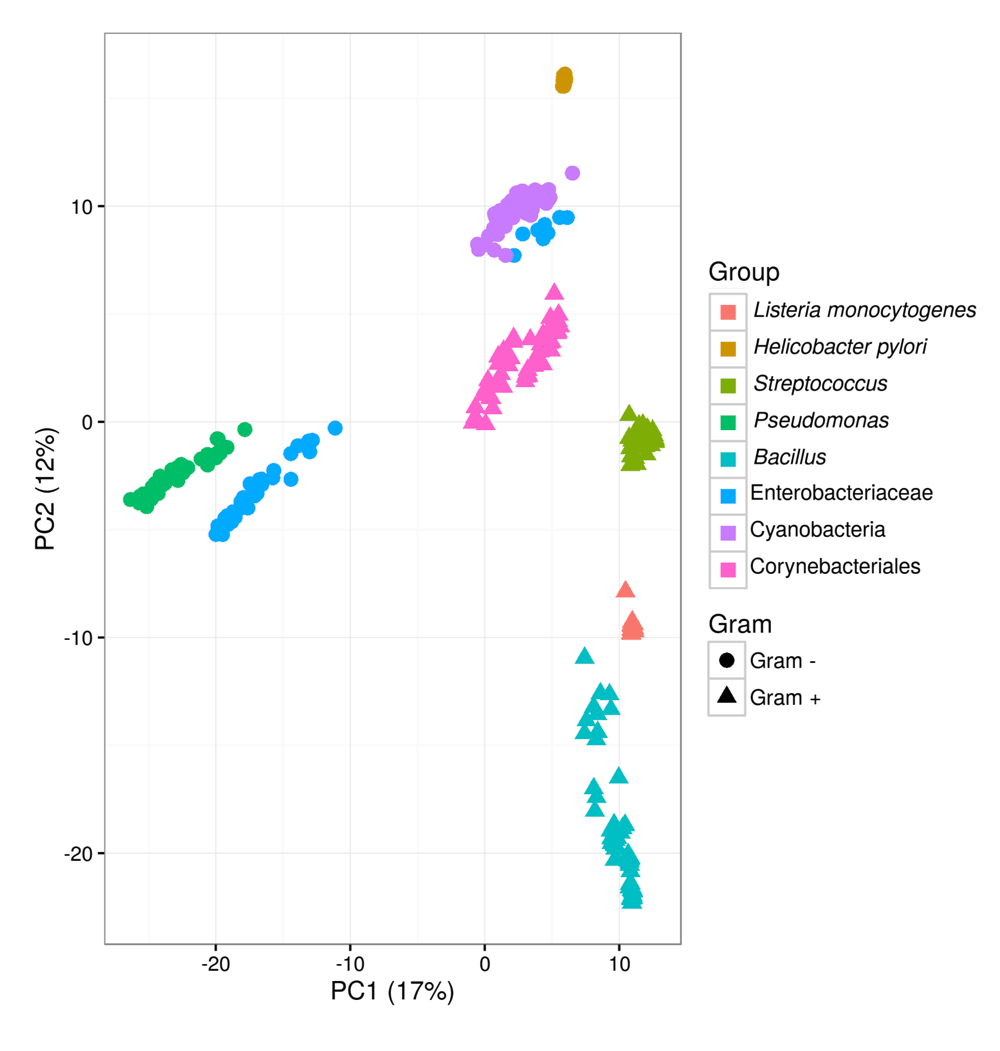
Principal component analysis of functional similarities of 446 genomes based on the presence/absence of domain architectures on the corresponding genomes. The variance explained by the first two components is indicated on axes labels.
DAB clusters can be labelled by their domain architecture and since this is a formal description of functional equivalence, results of independently obtained analyses can be combined. Figure 7 shows the results of a principal component analysis of the combined DAB clusters for selected genomes from eight taxa. The first two components account for a relatively low explained variance (29%) still grouping of genomes from the same taxa is apparent. High functional similarity among genomes of the same species (H. pylori and L. monocytogenes) is reflected by the compact clustering, while phylogenetically more distant genomes appear scattered in the functional space defined by the principal components.
We have shown that domain architecture-based methods can be used as an effective approach to identify clusters of functionally equivalent proteins, leading to results similar to those obtained by classical methods based on sequence similarity.
To assess whether DAB results were consistent with those of SB methods we chosen OrthaGogue as a representative of the latter class. Several tools such as COGNITOR39 and MultiPARANOID40 are available that implement different algorithm solutions to identify homologous sequences; however, despite different implementations, they all rely on sequence similarity as a proxy for functional equivalence. Here we considered SB methods as a golden standard for functional comparative genomics, especially when organisms within close evolutionary proximity are considered. Our aim was to investigate whether using domain architectures instead of sequence similarity alone would yield similar results, thereby justifying their use for large scale functional genome comparisons. Regarding domain architectures, we have explored different alternatives, as we have seen that the chosen database or set of reference domains plays a critical role; for example, the low coverage of TIGRFAM prevents the obtaining of reasonable clusters. The DAB approach takes advantage of the large computational effort that has already been devoted to the identification and definition of protein domains in dedicated databases such as Pfam. Protein domain models are built using large scale sequence comparisons which is an extremely computationally intensive task. However, once the domain models are defined, mining a sequence for domain occurrences is much less demanding task. Indeed, the task with the higher computational load (the definition of the domains) is performed only once and results can be stored and re-used for further analysis. This provides an effective scalable approach for large scale functional comparisons which by and large is independent of phylogenetic distances between species.
The chosen set of domain models and the database used as a reference greatly impact the results. InterPro aggregates protein domain signatures from different databases, which leads to redundancy of the domain models. This redundancy causes overlaps between the entries and an increase of the granularity of the clusters retrieved: this can bias downwards the size of the pan-genome and upwards the size of the core- genome, as shown in Table 1. In InterPro this redundancy is taken into account by implementing a hierarchy of protein families and domains. The entries at the top of these hierarchies correspond to broad families or domains that share higher level structure and/or function; the entries at the bottom correspond to specific functional subfamilies or structural /functional subclasses of domains30. Using Inter-Pro for DAB clustering would require taking into account the hierarchy of protein families and domains: however, this would pose challenges of its own and would require discrimination of the functional equivalence of different signatures within the same hierarchy.
Another source of redundancy are functionally equivalent domains from distantly related sequences. Pfam represents this through related families, termed clans, where relationships may be defined by similarity of sequence, structure or profile-HMM. Clans might contain functionally equivalent domains, however it is not clear whether this is always the case as the criteria for clan definition includes functional similarity but not functional equivalence41.
Members of a clan have diverging sequences and very often SB approaches would recognize the evolutionary distance between the sequences and group them in different clusters. If we were to assume that members of a clan are functionally equivalent and collect them in the same DA cluster, we will have a higher number of cases where a single DA cluster is split in multiple sequence clusters 1d→Ns. Also there would be higher number of cases of sequence clusters with the same DA but no exactly matching the DA clusters (1s→1d cases).
In many cases a one-to-one correspondence could be established between DAB and SB clusters indicating that often the sequence can be used as a proxy for function. At first this may seem a trivial result but it has a profound implication: domain model databases (in this case Pfam) contain enough information, encoded by known domain models, to represent the quasi totality of biological function encoded in the bacterial sequences analyzed here. However, it is important to stress that the comparisons have been performed considering sequences with known domains, representing currently around 85% of the genome coding content, a number that will only increase in the future.
A significant advantage of the DAB method over the SB method is that the domain architecture captured within a cluster can be used as a formal description of the function. Currently, more than 20% of all separable domains in the Pfam database, are so-called domains of unknown function (DUFs). Despite this, in bacterial species they are often essential42. With the DAB method they are formally included and often semantically linked to one or more domains of known function.
The starting position of the domains was used to generate labels indicating N-C terminal order of identified domains. The labels were used only for clustering as proteins sharing the same labels were assigned to the same clusters. Choosing instead the mid-point or the C-terminal position could affect the labeling but not the obtained clusters.
A content-wise formal labeling of DAB clusters makes a seamless integration of multiple independently performed DAB analysis possible. This allows for a comparison of potential functionomes across taxonomic boundaries, as presented in Figure 7, while new genomes can be added at a computational cost O(n), with n the number of genomes to be analyzed. SB methods that create orthologous groups require more memory and time as they come at an O(n2) computational cost. Other SB approaches, such as COGNITOR, reduce the computational costs to O(n) by using pre-computed databases. In this respect, the DAB approach is similar to the approach implemented in COGNITOR, by searching against existing databases of domains architectures. In this way the DAB approach leverages the extensive amount of work already put into defining domain families.
The bimodal shape of the distributions presented in Figure 3 and Figure 4 indicates the relative role of horizontal gene transfer and vertical descent when shaping bacterial genomes: the first peak accounts for sequences (or functions) only present in a small number of genome sequences which have been a likely acquired by horizontal gene transfer. The second peak accounts for high persistence genetic regions representing genes (or functions) belonging to the taxon core which have been likely acquired by vertical descent.
A measure of the impact of vertical descent and horizontal gene transfer is provided by the ratio between the core- and pan- genome sizes. The number of singletons provides a measure of the number of genes horizontally acquired from species outside the considered group.
Two of the most prominent differences between the two approaches are the number of retrieved singletons and the core- to pan-genome size ratio. Multiple members of the same taxon might acquire the same function through horizontal gene transfer43. This is likely to occur given that they would have similar physiological characteristics, hence they would tend to occupy a similar niche or, at least, more similar than when comparing species from different taxa. As the origin of the horizontally acquired genes may vary for each organism, an SB approach will correctly recognize the heterologous origin of the corresponding sequences and those will be assigned to singletons. However, the probabilistic hidden Markov models used for domain recognition are better at recognizing the functional similarity of the considered sequences and clusters them together.
Another indication of the relative impact of horizontal and vertical gene acquisition events is provided by the openness or closedness of the genome. Values for the decay parameter α in Table 3 indicate a relatively large impact of horizontal gene transfer. Within the considered taxa we observed αDAB > αSB, meaning that the sequence diversity is larger than the functional diversity: upon addition of new genomes to the sample the rate of addition of new sequence clusters appears higher than the rate of addition of new functions.
We have shown that domain architecture-based methods can be used as an effective approach to identify clusters of functionally equivalent proteins, leading to results similar to those obtained by classical methods based on sequence similarity. However, whether DAB methods are more accurate than SB methods to assess functional equivalence will require further analysis. In this light, results of functional conservation for both approaches could be compared in terms of GO similarity and/or EC number44,45. Partial domain hits might arise as a result of alignment, annotation and sequence assembly artefacts. To reduce the number of partial domain hits additional pruning could be implemented to distinguish these cases. However, this is an open problem that requires caution as it could influence the functional capacity of an organism and clustering approaches using DA.
The performance of DAB methods may be sub-optimal when dealing with newly sequenced genomes that are not yet well-characterized enough to have all of their domains present in domain databases, since DAB methods will be unable to handle unknown architectural types. Around 15% of the genome coding content corresponds to sequences with no identified protein domains. DAB approaches can be complemented with SB methods to consider these sequences or even protein sequences with low domain coverage, possible indicating the location of protein domains yet to be identified. Since DAB methods rely on the constant upgrading of public resources like UniProt and Pfam databases, an initial assessment of domain coverage appears as a sine qua non condition for application of these methods. DAB approaches could be used to assess the consistency of existing orthologous groups in terms of their domain architectures, at least when domain architectures are expected to be completely known in advance (for instance in the case of micro-evolutionary variations within a species where mutational events may disrupt a protein’s function). For other purposes, such as the discovery of a new phyla of cellular life that contains radically different domain architectures, global similarity methods may be preferred45.
As protein domain databases have evolved to the point where DAB and SB approaches produce similar results in closely related organisms, the DAB approach provides a fast and efficient alternative to SB methods to identify groups of functionally equivalent/related proteins for comparative genome analysis. The lower computational cost of DAB approaches makes them the better choice for large scale comparisons involving hundreds of genomes.
Highly redundant databases, such as InterPro, are best suited for domain based protein annotation, but are not effective for DAB clustering if the goal is to identify clusters of functionally equivalent proteins. To enable DAB approaches for highly structured databases, such as Inter-Pro, the hierarchy of protein families and domains within has to be explicitly considered. Currently Pfam is for this task a better alternative.
Differences between DAB and SB approaches increase when the goal is to study bacterial groups spanning wider evolutionary distances. The functional pan-genome is more closed in comparison to the sequence based pan-genome. Both methods have a distinct approach towards horizontally transferred genes, and the DAB approach has the potential to detect functional equivalence even when sequence similarities are low.
Complementing the standardly applied sequence similarity methods with a DAB approach pinpoints potential functional protein adaptations that may add to the overall fitness.
List of genomes used for the analysis at different phylogenetic levels. The genomes are grouped per taxonomic lineage used in this study.
Bacillus
GCA_000523045 Bacillus subtilis BEST7003
GCA_000782835 Bacillus subtilis
GCA_000832885 Bacillus thuringiensis str. Al Hakam
GCA_000473245 Bacillus infantis NRRL B-14911
GCA_000832585 Bacillus anthracis
GCA_000590455 Bacillus pumilus
GCA_000831065 Bacillus bombysepticus
GCA_000833275 Bacillus anthracis str. Turkey32
GCA_000952895 Bacillus sp.
GCA_000259365 Bacillus sp. JS
GCA_000143605 Bacillus cereus biovar anthracis str. CI
GCA_000186745 Bacillus subtilis BSn5
GCA_000987825 Bacillus methylotrophicus
GCA_000706725 Bacillus lehensis G1
GCA_000815145 Bacillus sp. Pc3
GCA_000496285 Bacillus toyonensis BCT-7112
GCA_000742855 Bacillus mycoides
GCA_000169195 Bacillus coagulans 36D1
GCA_000835145 Bacillus amyloliquefaciens KHG19
GCA_000321395 Bacillus subtilis subsp. subtilis str. BSP1
GCA_000009045 Bacillus subtilis subsp. subtilis str. 168
GCA_000293765 Bacillus subtilis QB928
GCA_000025805 Bacillus megaterium DSM 319
GCA_000747345 Bacillus sp. X1(2014)
GCA_000833005 Bacillus amyloliquefaciens
GCA_000408885 Bacillus paralicheniformis ATCC 9945a
GCA_000742895 Bacillus anthracis str. Vollum
GCA_000829195 Bacillus sp. OxB-1
GCA_000800825 Bacillus sp. WP8
GCA_000706705 Bacillus subtilis subsp. subtilis str. OH 131.1
GCA_000338735 Bacillus subtilis XF-1
GCA_000832445 Bacillus anthracis
GCA_000747335 Bacillus anthracis
GCA_000008505 Bacillus thuringiensis serovar konkukian str. 97-27
GCA_000195515 Bacillus amyloliquefaciens TA208
GCA_000209795 Bacillus subtilis subsp. natto BEST195
GCA_000017425 Bacillus cytotoxicus NVH 391-98
GCA_000877815 Bacillus sp. YP1
GCA_000177235 Bacillus cellulosilyticus DSM 2522
GCA_000344745 Bacillus subtilis subsp. subtilis 6051-HGW
GCA_000227485 Bacillus subtilis subsp. subtilis str. RO-NN-1
GCA_000494835 Bacillus amyloliquefaciens CC178
GCA_000011145 Bacillus halodurans C-125
GCA_000724485 Bacillus methanolicus MGA3
GCA_000018825 Bacillus weihenstephanensis KBAB4
GCA_000005825 Bacillus pseudofirmus OF4
GCA_000017885 Bacillus pumilus SAFR-032
GCA_000583065 Bacillus methylotrophicus Trigo-Cor1448
GCA_000349795 Bacillus subtilis subsp. subtilis str. BAB-1
GCA_000306745 Bacillus thuringiensis Bt407
GCA_000011645 Bacillus licheniformis DSM 13 = ATCC 14580
GCA_000497485 Bacillus subtilis PY79
GCA_000009825 Bacillus clausii KSM-K16
GCA_000227465 Bacillus subtilis subsp. spizizenii TU-B-10
GCA_000971925 Bacillus subtilis KCTC 1028
GCA_000972245 Bacillus endophyticus
GCA_000242895 Bacillus sp. 1NLA3E
GCA_000832485 Bacillus thuringiensis
GCA_000830075 Bacillus atrophaeus
GCA_000146565 Bacillus subtilis subsp. spizizenii str. W23
Corynebacteriales
GCA_000016005 Mycobacterium sp. JLS
GCA_000758405 Mycobacterium abscessus subsp. bolletii
GCA_000283295 Mycobacterium smegmatis str. MC2 155
GCA_001021045 Corynebacterium testudinoris
GCA_000341345 Corynebacterium halotolerans YIM 70093 = DSM 44683
GCA_000525655 Corynebacterium falsenii DSM 44353
GCA_000255195 Corynebacterium diphtheriae HC04
GCA_000523235 Nocardia nova SH22a
GCA_000026685 Mycobacterium leprae Br4923
GCA_000980815 Corynebacterium camporealensis
GCA_000328565 Mycobacterium sp. JS623
GCA_000015405 Mycobacterium sp. KMS
GCA_000987865 [Brevibacterium] flavum
GCA_001020985 Corynebacterium mustelae
GCA_001021065 Corynebacterium uterequi
GCA_000177535 Corynebacterium resistens DSM 45100
GCA_000011305 Corynebacterium efficiens YS-314
GCA_000835265 Mycobacterium avium subsp. paratuberculosis
GCA_000739455 Corynebacterium imitans
GCA_000831265 Mycobacterium kansasii 662
GCA_000819445 Corynebacterium humireducens NBRC 106098 = DSM 45392
GCA_000770235 Mycobacterium avium subsp. avium
GCA_000980835 Corynebacterium kutscheri
GCA_000010225 Corynebacterium glutamicum R
GCA_000590555 Corynebacterium argentoratense DSM 44202
GCA_000247715 Gordonia polyisoprenivorans VH2
GCA_000416365 Mycobacterium sp. VKM Ac-1817D
GCA_000418365 Corynebacterium terpenotabidum Y-11
GCA_000092225 Tsukamurella paurometabola DSM 20162
GCA_000442645 Corynebacterium maris DSM 45190
GCA_000277125 Mycobacterium intracellulare ATCC 13950
GCA_000196695 Rhodococcus equi 103S
GCA_000828995 Mycobacterium tuberculosis str. Kurono
GCA_000006605 Corynebacterium jeikeium K411
GCA_000022905 Corynebacterium aurimucosum
GCA_001021025 Corynebacterium epidermidicanis
GCA_000010105 Rhodococcus erythropolis PR4
GCA_000092825 Segniliparus rotundus DSM 44985
GCA_000758245 Mycobacterium bovis
GCA_000184435 Mycobacterium gilvum Spyr1
GCA_000829075 Mycobacterium avium subsp. hominissuis TH135
GCA_000214175 Amycolicicoccus subflavus DQS3-9A1
GCA_000769635 Corynebacterium ulcerans
GCA_000626675 Corynebacterium glyciniphilum AJ 3170
GCA_001026945 Corynebacterium pseudotuberculosis
GCA_000026445 Mycobacterium liflandii 128FXT
GCA_000013925 Mycobacterium ulcerans Agy99
GCA_000954115 Rhodococcus sp. B7740
GCA_000143885 Gordonia sp. KTR9
GCA_000014565 Rhodococcus jostii RHA1
GCA_000179395 Corynebacterium variabile DSM 44702
GCA_000732945 Corynebacterium atypicum
GCA_000723425 Mycobacterium marinum E11
GCA_000230895 Mycobacterium rhodesiae NBB3
GCA_000344785 Corynebacterium callunae DSM 20147
GCA_000010805 Rhodococcus opacus B4
GCA_000982715 Rhodococcus aetherivorans
GCA_000298095 Mycobacterium indicus pranii MTCC 9506
GCA_000833575 Corynebacterium singulare
GCA_000023145 Corynebacterium kroppenstedtii DSM 44385
Cyanobacteria
GCA_000317085 Synechococcus sp. PCC 7502
GCA_000011385 Gloeobacter violaceus PCC 7421
GCA_000014585 Synechococcus sp. CC9311
GCA_000012465 Prochlorococcus marinus str. NATL2A
GCA_000737535 Synechococcus sp. KORDI-100
GCA_000013205 Synechococcus sp. JA-3-3Ab
GCA_000021825 Cyanothece sp. PCC 7424
GCA_000063505 Synechococcus sp. WH 7803
GCA_000022045 Cyanothece sp. PCC 7425
GCA_000316575 Calothrix sp. PCC 7507
GCA_000316685 Synechococcus sp. PCC 6312
GCA_000012505 Synechococcus sp. CC9902
GCA_000317475 Oscillatoria nigro-viridis PCC 7112
GCA_000063525 Synechococcus sp. RCC307
GCA_000317695 Anabaena cylindrica PCC 7122
GCA_000014265 Trichodesmium erythraeum IMS101
GCA_000817325 Synechococcus sp. UTEX 2973
GCA_000737575 Synechococcus sp. KORDI-49
GCA_000317125 Chroococcidiopsis thermalis PCC 7203
GCA_000017845 Cyanothece sp. ATCC 51142
GCA_000020025 Nostoc punctiforme PCC 73102
GCA_000018105 Acaryochloris marina MBIC11017
GCA_000757865 Prochlorococcus sp. MIT 0801
GCA_000317045 Geitlerinema sp. PCC 7407
GCA_000012625 Synechococcus sp. CC9605
GCA_000737595 Synechococcus sp. KORDI-52
GCA_000317635 Halothece sp. PCC 7418
GCA_000025125 Candidatus Atelocyanobacterium thalassa isolate ALOHA
GCA_000010625 Microcystis aeruginosa NIES-843
GCA_000317065 Pseudanabaena sp. PCC 7367
GCA_000312705 Anabaena sp. 90
GCA_000316515 Cyanobium gracile PCC 6307
GCA_000316605 Leptolyngbya sp. PCC 7376
GCA_000317025 Pleurocapsa sp. PCC 7327
GCA_000009705 Nostoc sp. PCC 7120
GCA_000013225 Synechococcus sp. JA-2-3B’a(2–13)
GCA_000757845 Prochlorococcus sp. MIT 0604
GCA_000317515 Microcoleus sp. PCC 7113
GCA_000734895 Calothrix sp. 336/3
GCA_000007925 Prochlorococcus marinus subsp. marinus str. CCMP1375
GCA_000021805 Cyanothece sp. PCC 8801
GCA_000019485 Synechococcus sp. PCC 7002
GCA_000317655 Cyanobacterium stanieri PCC 7202
GCA_000316625 Nostoc sp. PCC 7107
GCA_000011465 Prochlorococcus marinus subsp. pastoris str. CCMP1986
GCA_000316665 Rivularia sp. PCC 7116
GCA_000317105 Oscillatoria acuminata PCC 6304
GCA_000317435 Calothrix sp. PCC 6303
GCA_000317555 Gloeocapsa sp. PCC 7428
GCA_000478825 Synechocystis sp. PCC 6714
GCA_000204075 Anabaena variabilis ATCC 29413
GCA_000317575 Stanieria cyanosphaera PCC 7437
GCA_000161795 Synechococcus sp. WH 8109
GCA_000011345 Thermosynechococcus elongatus BP-1
GCA_000317615 Dactylococcopsis salina PCC 8305
GCA_000284135 Synechocystis sp. PCC 6803 substr. GT-I
GCA_000024045 Cyanothece sp. PCC 8802
GCA_000317495 Crinalium epipsammum PCC 9333
GCA_000317675 Cyanobacterium aponinum PCC 10605
GCA_000012525 Synechococcus elongatus PCC 7942
Enterobacteriaceae
GCA_000259175 Providencia stuartii MRSN 2154
GCA_000214805 Serratia sp. AS13
GCA_000330865 Serratia marcescens FGI94
GCA_001010285 Photorhabdus temperata subsp. thracensis
GCA_000364725 Candidatus Moranella endobia PCVAL
GCA_000521525 Buchnera aphidicola str. USDA (Myzus persicae)
GCA_000517405 Candidatus Sodalis pierantonius str. SOPE
GCA_000012005 Shigella dysenteriae Sd197
GCA_000196475 Photorhabdus asymbiotica
GCA_000750295 Salmonella enterica subsp. enterica serovar Enteritidis
GCA_000007885 Yersinia pestis biovar Microtus str. 91001
GCA_000739495 Klebsiella pneumoniae
GCA_000252995 Salmonella bongori NCTC 12419
GCA_000270125 Pantoea ananatis AJ13355
GCA_000215745 Enterobacter aerogenes KCTC 2190
GCA_000092525 Shigella sonnei Ss046
GCA_000020865 Edwardsiella tarda EIB202
GCA_000023545 Dickeya dadantii Ech703
GCA_000238975 Serratia symbiotica str. ’Cinara cedri’
GCA_000975245 Serratia liquefaciens
GCA_000006645 Yersinia pestis KIM10+
GCA_000224675 Enterobacter asburiae LF7a
GCA_000007405 Shigella flexneri 2a str. 2457T
GCA_001022275 Citrobacter freundii
GCA_000963575 Klebsiella michiganensis
GCA_000504545 Cronobacter sakazakii CMCC 45402
GCA_000012025 Shigella boydii Sb227
GCA_000814125 Enterobacter cloacae
GCA_000987925 Yersinia enterocolitica
GCA_000011745 Candidatus Blochmannia pennsylvanicus str. BPEN
GCA_000255535 Rahnella aquatilis HX2
GCA_000952955 Escherichia coli
GCA_000695995 Serratia sp. FS14
GCA_000648515 Citrobacter freundii CFNIH1
GCA_001022295 Klebsiella oxytoca
GCA_000147055 Dickeya dadantii 3937
GCA_000348565 Edwardsiella piscicida C07-087
GCA_000742755 Klebsiella pneumoniae subsp. pneumoniae
GCA_000027225 Xenorhabdus bovienii SS-2004
GCA_000247565 Wigglesworthia glossinidia endosymbiont of Glossina morsitans morsitans (Yale colony)
GCA_000828815 Candidatus Tachikawaea gelatinosa
GCA_000022805 Yersinia pestis D106004
GCA_001006005 Serratia fonticola
GCA_000018625 Salmonella enterica subsp. arizonae serovar 62:z4,z23:-
GCA_000478905 Candidatus Pantoea carbekii
GCA_000410515 Enterobacter sp. R4-368
GCA_000148935 Pantoea vagans C9-1
GCA_000444425 Proteus mirabilis BB2000
GCA_000747565 Serratia sp. SCBI
GCA_001022135 Kluyvera intermedia
GCA_000757825 Cedecea neteri
GCA_000294535 Pectobacterium carotovorum subsp. carotovorum PCC21
GCA_000834375 Yersinia pseudotuberculosis YPIII
GCA_000043285 Candidatus Blochmannia floridanus
GCA_000093065 Candidatus Riesia pediculicola USDA
GCA_000834515 Yersinia intermedia
GCA_000759475 Pantoea rwandensis
GCA_000027065 Siccibacter turicensis z3032
GCA_000582515 Yersinia similis
GCA_000300455 Kosakonia sacchari SP1
Helicobacter pylori
GCA_000148855 Helicobacter pylori SJM180
GCA_000021165 Helicobacter pylori G27
GCA_000185245 Helicobacter pylori SouthAfrica7
GCA_000093185 Helicobacter pylori v225d
GCA_000277365 Helicobacter pylori Shi417
GCA_000498315 Helicobacter pylori BM012A
GCA_000270065 Helicobacter pylori F57
GCA_000392455 Helicobacter pylori UM032
GCA_000277385 Helicobacter pylori Shi169
GCA_000008525 Helicobacter pylori 26695
GCA_000270045 Helicobacter pylori F32
GCA_000148915 Helicobacter pylori Sat464
GCA_000185225 Helicobacter pylori Lithuania75
GCA_000600045 Helicobacter pylori oki102
GCA_000600205 Helicobacter pylori oki828
GCA_000192335 Helicobacter pylori 2018
GCA_000827025 Helicobacter pylori
GCA_000590775 Helicobacter pylori SouthAfrica20
GCA_000270025 Helicobacter pylori F30
GCA_000148665 Helicobacter pylori 908
GCA_000392515 Helicobacter pylori UM037
GCA_000392475 Helicobacter pylori UM299
GCA_000262655 Helicobacter pylori XZ274
GCA_000008785 Helicobacter pylori J99
GCA_000685745 Helicobacter pylori
GCA_000185205 Helicobacter pylori Gambia94/24
GCA_000826985 Helicobacter pylori 26695-1
GCA_000315955 Helicobacter pylori Aklavik117
GCA_000498335 Helicobacter pylori BM012S
GCA_000277405 Helicobacter pylori Shi112
GCA_000224535 Helicobacter pylori Puno120
GCA_000317875 Helicobacter pylori Aklavik86
GCA_000600185 Helicobacter pylori oki673
GCA_000196755 Helicobacter pylori B8
GCA_000439295 Helicobacter pylori UM298
GCA_000348885 Helicobacter pylori OK310
GCA_000307795 Helicobacter pylori 26695
GCA_000013245 Helicobacter pylori HPAG1
GCA_000392535 Helicobacter pylori UM066
GCA_000185185 Helicobacter pylori India7
GCA_000213135 Helicobacter pylori 83
GCA_000685705 Helicobacter pylori
GCA_000224575 Helicobacter pylori SNT49
GCA_000600085 Helicobacter pylori oki112
GCA_000023805 Helicobacter pylori 52
GCA_000348865 Helicobacter pylori OK113
GCA_000259235 Helicobacter pylori HUP-B14
GCA_000020245 Helicobacter pylori Shi470
GCA_000270005 Helicobacter pylori F16
GCA_000192315 Helicobacter pylori 2017
GCA_000685665 Helicobacter pylori
GCA_000600165 Helicobacter pylori oki422
GCA_000255955 Helicobacter pylori ELS37
GCA_000021465 Helicobacter pylori P12
GCA_000600145 Helicobacter pylori oki154
GCA_000224555 Helicobacter pylori Puno135
GCA_000011725 Helicobacter pylori 51
GCA_000148895 Helicobacter pylori Cuz20
GCA_000817025 Helicobacter pylori
GCA_000178935 Helicobacter pylori 35A
Listeria monocytogenes
GCA_000438745 Listeria monocytogenes
GCA_000438705 Listeria monocytogenes
GCA_001027125 Listeria monocytogenes
GCA_000438725 Listeria monocytogenes
GCA_000197755 Listeria monocytogenes
GCA_001027245 Listeria monocytogenes
GCA_001027085 Listeria monocytogenes
GCA_001005925 Listeria monocytogenes
GCA_000746625 Listeria monocytogenes
GCA_000382925 Listeria monocytogenes
GCA_000438665 Listeria monocytogenes
GCA_000800335 Listeria monocytogenes
GCA_001027165 Listeria monocytogenes
GCA_000438605 Listeria monocytogenes
GCA_000438585 Listeria monocytogenes
GCA_000808055 Listeria monocytogenes
GCA_000950775 Listeria monocytogenes
GCA_001027065 Listeria monocytogenes
GCA_000600015 Listeria monocytogenes
GCA_001027205 Listeria monocytogenes
GCA_000438685 Listeria monocytogenes
GCA_001005985 Listeria monocytogenes
GCA_000438625 Listeria monocytogenes
GCA_000681515 Listeria monocytogenes
GCA_000438645 Listeria monocytogenes
GCA_000210815 Listeria monocytogenes
Pseudomonas
GCA_000829885 Pseudomonas aeruginosa
GCA_000510285 Pseudomonas monteilii SB3078
GCA_000988485 Pseudomonas syringae pv. syringae B301D
GCA_000013785 Pseudomonas stutzeri A1501
GCA_000759535 Pseudomonas cremoricolorata
GCA_000953455 Pseudomonas pseudoalcaligenes
GCA_000981825 Pseudomonas aeruginosa
GCA_000661915 Pseudomonas stutzeri
GCA_000508205 Pseudomonas sp. TKP
GCA_000014625 Pseudomonas aeruginosa UCBPP-PA14
GCA_000019445 Pseudomonas putida W619
GCA_000316175 Pseudomonas sp. UW4
GCA_000498975 Pseudomonas mosselii SJ10
GCA_000473745 Pseudomonas aeruginosa VRFPA04
GCA_000691565 Pseudomonas putida
GCA_000730425 Pseudomonas fluorescens
GCA_000007805 Pseudomonas syringae pv. tomato str. DC3000
GCA_000349845 Pseudomonas denitrificans ATCC 13867
GCA_000026105 Pseudomonas entomophila L48
GCA_000689415 Pseudomonas knackmussii B13
GCA_000325725 Pseudomonas putida HB3267
GCA_000412695 Pseudomonas resinovorans NBRC 106553
GCA_000831585 Pseudomonas plecoglossicida
GCA_000756775 Pseudomonas sp. 20_BN
GCA_000590475 Pseudomonas stutzeri
GCA_000829255 Pseudomonas aeruginosa
GCA_000761155 Pseudomonas rhizosphaerae
GCA_001038645 Pseudomonas stutzeri
GCA_000264665 Pseudomonas putida ND6
GCA_000007565 Pseudomonas putida KT2440
GCA_000494915 Pseudomonas sp. VLB120
GCA_000226155 Pseudomonas aeruginosa M18
GCA_000213805 Pseudomonas fulva 12-X
GCA_000194805 Pseudomonas brassicacearum subsp. brassicacearum NFM421
GCA_000336465 Pseudomonas poae RE*1-1-14
GCA_000828695 Pseudomonas protegens Cab57
GCA_000800255 Pseudomonas parafulva
GCA_000257545 Pseudomonas mandelii JR-1
GCA_000012205 Pseudomonas savastanoi pv. phaseolicola 1448A
GCA_000816985 Pseudomonas aeruginosa
GCA_000746525 Pseudomonas alkylphenolia
GCA_000496605 Pseudomonas aeruginosa PA1
GCA_000204295 Pseudomonas mendocina NK-01
GCA_000829415 Pseudomonas sp. StFLB209
GCA_000012265 Pseudomonas protegens Pf-5
GCA_000412675 Pseudomonas putida NBRC 14164
GCA_000397205 Pseudomonas protegens CHA0
GCA_000648735 Pseudomonas syringae pv. actinidiae ICMP 18884
GCA_000012245 Pseudomonas syringae pv. syringae B728a
GCA_000761195 Pseudomonas chlororaphis subsp. aurantiaca
GCA_000818015 Pseudomonas balearica DSM 6083
GCA_000219605 Pseudomonas stutzeri ATCC 17588 = LMG 11199
GCA_000219705 Pseudomonas putida S16
GCA_000511325 Pseudomonas sp. FGI182
GCA_000508765 Pseudomonas aeruginosa LES431
GCA_000297075 Pseudomonas pseudoalcaligenes CECT 5344
GCA_000517305 Pseudomonas cichorii JBC1
GCA_000963835 Pseudomonas chlororaphis
GCA_000327065 Pseudomonas stutzeri RCH2
GCA_000271365 Pseudomonas aeruginosa DK2
Streptococcus
GCA_000211015 Streptococcus pneumoniae SPN034183
GCA_000210975 Streptococcus pneumoniae INV104
GCA_000203195 Streptococcus gallolyticus subsp. gallolyticus ATCC BAA-2069
GCA_001020185 Streptococcus pyogenes
GCA_000253155 Streptococcus oralis Uo5
GCA_000696505 Streptococcus equi subsp. zooepidemicus CY
GCA_000463355 Streptococcus intermedius B196
GCA_000698885 Streptococcus thermophilus ASCC 1275
GCA_000014205 Streptococcus sanguinis SK36
GCA_000007045 Streptococcus pneumoniae R6
GCA_000306805 Streptococcus intermedius JTH08
GCA_000196595 Streptococcus pneumoniae TCH8431/19A
GCA_000262145 Streptococcus parasanguinis FW213
GCA_001026925 Streptococcus agalactiae
GCA_000251085 Streptococcus pneumoniae ST556
GCA_000019025 Streptococcus pneumoniae Taiwan19F-14
GCA_000211055 Streptococcus pneumoniae SPN994039
GCA_000688775 Streptococcus sp. VT 162
GCA_000231905 Streptococcus suis D12
GCA_000026665 Streptococcus pneumoniae ATCC 700669
GCA_000283635 Streptococcus macedonicus ACA-DC 198
GCA_000014365 Streptococcus pneumoniae D39
GCA_000019265 Streptococcus pneumoniae Hungary19A-6
GCA_000299015 Streptococcus pneumoniae gamPNI0373
GCA_000019985 Streptococcus pneumoniae CGSP14
GCA_000463395 Streptococcus constellatus subsp. pharyngis C232
GCA_000187935 Streptococcus parauberis NCFD 2020
GCA_000253315 Streptococcus salivarius JIM8777
GCA_000427055 Streptococcus agalactiae ILRI112
GCA_000246835 Streptococcus infantarius subsp. infantarius CJ18
GCA_000427075 Streptococcus agalactiae ILRI005
GCA_000007465 Streptococcus mutans UA159
GCA_000831165 Streptococcus anginosus
GCA_000147095 Streptococcus pneumoniae 670-6B
GCA_000817005 Streptococcus pneumoniae
GCA_000180515 Streptococcus pneumoniae SPNA45
GCA_000441535 Streptococcus lutetiensis 033
GCA_000210955 Streptococcus pneumoniae OXC141
GCA_000009545 Streptococcus uberis 0140J
GCA_000648555 Streptococcus iniae
GCA_000027165 Streptococcus mitis B6
GCA_000018985 Streptococcus pneumoniae JJA
GCA_000270165 Streptococcus pasteurianus ATCC 43144
GCA_000479315 Streptococcus sp. I-P16
GCA_000478925 Streptococcus anginosus subsp. whileyi MAS624
GCA_000019825 Streptococcus pneumoniae G54
GCA_000017005 Streptococcus gordonii str. Challis substr. CH1
GCA_000479335 Streptococcus sp. I-G2
GCA_000385925 Streptococcus oligofermentans AS 1.3089
GCA_000210935 Streptococcus pneumoniae INV200
GCA_000211035 Streptococcus pneumoniae SPN994038
GCA_000221985 Streptococcus pseudopneumoniae IS7493
GCA_000006885 Streptococcus pneumoniae TIGR4
GCA_000018965 Streptococcus pneumoniae 70585
GCA_000348705 Streptococcus pneumoniae PCS8235
GCA_000210995 Streptococcus pneumoniae SPN034156
GCA_000231925 Streptococcus suis ST1
GCA_000019005 Streptococcus pneumoniae P1031
GCA_000188715 Streptococcus dysgalactiae subsp. equisimilis ATCC 12394
GCA_000026585 Streptococcus equi subsp. equi 4047
JJK, MSD, ES, PJS participated in the set-up of the research. JJK and MSD were responsible for the analysis. JJK, ES, PJS, MSD and VdS wrote the manuscript. All authors critically revised the manuscript.
This work was partly supported by the European Union’s Horizon 2020 research and innovation programme (EmPowerPutida, Contract No. 635536, granted to Vitor A P Martins dos Santos).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.

Cluster persistence is defined as the relative number of genomes with at least one protein assigned to the cluster. The plots show frequency of SB clusters according to their persistence. Publicly available and complete genome sequences assigned to each taxon were selected so that phylogenetic diversity within the taxon was preserved, as described in materials and methods. 60 distinct genome sequences were considered for each taxon shown.

The plots show frequency of DAB clusters according to their persistence.

On the left DAB is used as a reference and each bar represents the relative frequency of one DAB cluster containing sequences assigned to {1, 2, . . . , 5} and 6 or more SB clusters and one-to-one represents the relative frequency of identical cluster. Similarly, on the right SB is used as a reference. Axis labels follow notation in Figure 5
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)