Keywords
Ebolavirus, LCK, HLA, viruses, T-cells, GEO, pathways, pathogenesis
This article is included in the Emerging Diseases and Outbreaks gateway.
This article is included in the Ebola Virus collection.
Ebolavirus, LCK, HLA, viruses, T-cells, GEO, pathways, pathogenesis
Ebola virus (EBOV), a known member of filoviruses, causes hemorrhagic fever related epidemics, resulting in disease severity and high case fatality rates (Sanchez et al., 2006). The severity of the illness is an end result of pathogenetic and complex mechanisms exploited by the virus to suppress immune responses, both innate and adaptive (Hoenen et al., 2006). EBOV remains an important threat to public health, specifically in central Africa, but also worldwide, due to imported infections and fear of biological terrorism misuse. Of the four known EBOV species (Zaire, Sudan, Cote d’Ivoire and Reston), only Zaire and Sudan EBOVs species have resulted in large epidemics with intense hemorrhagic fever (Johnson et al., 1977; Georges et al., 1999). Due to several outbreaks and high fatality rates recorded in many Zaire EBOV cases, this species is the most extensively studied (Mahanty & Bray, 2004). Infections from EBOV induce a response to systemic inflammation resulting in dysfunctional patterns in blood clotting, and vascular and immune systems. This systemic imbalance leads to failure in several body organs, which is a similar phenomenon to septic shock (Achinko & Dormer, 2015). Fatalities resulting from EBOV infections, combined with the lack of treatment and approved vaccines, keeps EBOV on the list of most important public health and category A bio-threat pathogens (Hoenen et al., 2006). Although a lot of work is currently ongoing to identify the right genetic mechanisms exploited by the virus in its host leading to pathogenesis, little is currently known of its pathogenetic pathway.
Enveloped EBOV particles consist of an RNA genome, which is negatively stranded and non-segmented. The fourth gene of the linear genome encodes different and specific glycoproteins, with the precursor for the viral soluble nonstructural glycoprotein (pre-sGP) known as the main product of this gene. After posttranslational cleavage of furin molecules, pre-sGP matures to sGP and delta peptide, which are both secreted from infected cells, with sGP also identified in the serum of infected individuals (Sanchez et al., 1996; Volchkova et al., 1998). Among the Mononegavirales, EBOV is the only virus to produce a soluble glycoprotein (GP) (Volchkova et al., 1998). sGP has an associated role with viral pathogenesis, due to its relative abundance in circulation and the presence of non-infected B- and T-cells subjected to intense bystander apoptosis through a previously unknown mechanism, which suggests a possible interaction between sGP and important related immune cells (Baize et al., 2000; Geisbert et al., 2000). An attempt by Wolf et al. (2011) to prove this concept using recombinant sGP alone or together with death receptors (tumor necrosis factor-related apoptosis-inducing ligand and FAS) showed neither a decrease or increase in apoptosis in Jurkat cells (a human lymphocytic cell line), exempting it from bystander apoptosis induction.
The known EBOV entry model leading to infection occurs through minute skin and mucosa lesions of the host, further infecting macrophages and dendritic cells (DCs) as the main target cells (Schnittler & Feldmann, 1998). In mice, macrophages are primary targets (Bray et al., 1998; Gibb et al., 2001) and may be infected as early as two days post infection in non-human primates (Geisbert et al., 2003; Ryabchikova et al., 1999). Several protein molecules may play a role in the cellular attachment for EBOV and they include: b1-integrin receptor, DC specific C-type lectins or lymph node/liver specific (L) intercellular adhesion molecule 3 grabbing nonintegrin (DC-SIGN and L-SIGN, respectively; CD209), C-type lectin of the sinusoidal cells of liver and lymph nodes, C-type lectin with galactose and N-acetylgalactosamine specific to human macrophages, and factors related to DC-SIGN (Hensley et al., 2005). During an innate immune response from EBOV infection, the interferon (IFN) response is considered very critical for disease outcome in mice infected by EBOV-wild-type (Bray, 2001), and mice will die within a week when treated with anti-IFN antibodies. Mice that lack receptors for INF-a/b or signal transducer and activator of transcription-1 (STAT-1), which are necessary for IFN signaling, are susceptible to EBOV infection (Bray, 2001). The block of IFN signaling was also associated with p38 phosphorylation inhibition, a central molecule for the mitogen activated-protein-kinase (MAPK) signaling pathway linked to IFN. Comparatively, DCs, which are crucially involved with both innate and adaptive immunity, don’t seem to properly execute their function post-EBOV infection. In vitro demonstrations showed that DCs infected with the virus failed to generate pro-inflammatory cytokines and expression of co-stimulatory molecules, like CD80 or CD86, resulting in the absence of T-cell proliferation and abnormal maturation (Bosio et al., 2003; Mahanty et al., 2003). EBOV infection in humans shows evidence of intravascular apoptosis and loss of mRNA for CD3, CD8 and T-cell receptor (TCR)-Vb from peripheral blood mononuclear cells (Baize et al., 1999).
From our last study (Achinko & Dormer, 2015), we showed that the Lymphocyte Tyrosine Kinase (LCK) was dominantly associated with immune related functions in an EBOV and host protein interaction map. With this information, more studies on LCK guided our understanding in the role it plays in EBOV infection. LCK is involved in a pathway for TCR activation (Palacios & Weiss, 2004) and is dominantly expressed in T-cells with localization in microdomains. It plays crucial roles in several immune related processes, such as activation of early immune events, cell cycle progression, differentiation of the thymus and Th1/Th2 cells (Yamashita et al., 1998), homeostatic proliferation and apoptosis of naïve T-cells. LCK is organized in domains: a C-terminal kinase domain, homology domains to Src (SH4, SH3 and SH2) and the N-terminal region. SH3 is important in protein-protein interactions and recruits downstream molecules like phosphatidylinositol-4,5-biphosphate 3-kinase catalytic subunit (PI3K) and MAPK (Togni et al., 2004). LCK protein expression significantly decreases in different types of cancers (Majolini et al., 1999) and type 1 diabetes (Nervi et al., 2002), but it has not been associated with chronic viral related diseases like EBOV. The human LCK gene is 14kb in size with 12 exons, implicating it with alternative splicing events (Modrek et al., 2001). Two structurally different promoter regions (distal and proximal) separated by ~35kb, control the gene (Takadera et al., 1989). Through differential usage of each promoter, type I mRNA is produced from the proximal promoter and type II mRNA from the distal promoter. Alternative splicing at the distal promoter with about five different start sites, produces three different mRNAs, two of which are dominantly oncogenic related, leading to a loss of interaction with CD4 and CD8 (Huse et al., 1998), and CD3/TCR (Goldman et al., 1998). Interleukin (IL)2 is a T-cell growth factor and is known to induce human peripheral T-cell lymphocytes to re-enter the cell cycle. Due to a lack of intrinsic catalytic activity at the IL2 receptor, early responses to IL2 stimulation originate from cytoplasmic enzymes with receptor association. It has been shown in peripheral blood lymphocytes that IL2 is stimulated through a dependent tyrosine phosphorylation pattern engaging the LCK SH2 domain (Vogel & Fujita, 1995).
The associated evidence of LCK with EBOV and immune activation prompted the design of this study by exploring data from the Gene Expression Omnibus (GEO) database (Wilhite & Barrett, 2012) hosted at the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/), which was created in 2000 for storing high throughput gene expression data, exploited for research and dissemination (Edgar et al., 2002). Functional genomics datasets deposited here are both from microarray experiments and sequence-based technologies (Wilhite & Barrett, 2012).
The only EBOV data documented in GEO is from Zaire species. Through NCBI Entrez Query (Sayers et al., 2009), related EBOV data, with humans as hosts, was obtained by the present study and the relative mean (RMEAN) values of genes per sample were used to perform expression variation analysis across datsets, which grouped samples in three clusters based on LCK and/or CD209 expression. All samples showed a low expression of major histocompatibility complex (MHC) class I human leukocyte antigen (HLA) immune genes, which are critical for viral clearance. This study brings to light a possible immune pathway exploited by EBOV and other related viruses for T-cell infection and immune suppression. Given the need for sequenced data on various EBOV species to enable intra species comparative analysis, our group is currently trying to decipher a possible EBOV immune pathway, with implications for identifying potent antiviral therapeutic targets and possible markers to differentiate viral disease.
Based on our previous publication on differential immune responses on relative antigenic surfaces presented by EBOV (Achinko & Dormer, 2015), we aimed to understand the variation in viral envelope GP surfaces and their post signaling responses within the cell. Data was extracted from GEO (RRID: SCR_005012), which has three divisions: a) GEO Database (www.ncbi.nlm.nih.gov/geo/), a public repository which provides tools to query functional genomics data and download experiments and gene expression curated profiles from deposited sequenced and micro-array datasets; b) GEO Datasets (www.ncbi.nlm.nih.gov/gds), a storage site for molecular abundance and curated gene expression datasets assembled from the GEO repository; and c) GEO Profiles (www.ncbi.nlm.nih.gov/geoprofiles/), a storage site for individual molecular abundance and gene expression profiles assembled from the GEO repository. We focused on GEO Profiles, and using the terms “GP (viral Envelope glycoprotein)” and “virus”, all data related to experiments targeting these words, including EBOV, was extracted for further analysis.
The data obtained per sample had the following parameters: sample identity, title of the study, relative standard deviation (RSTD), relative mean (RMEAN), gene name, Genebank accession number, taxon and GEO dataset type (Dataset 1; Achinko et al., 2016a). Genes of Interest, with signaling as their molecular function and previously shown to be dominantly involved in an EBOV gene interaction map (Achinko & Dormer, 2015) (MVP, POLG, HLA, SRC, LCK, MERTK, TYRO3, UFO, ACK1, BMPR2, YES, FYN, ROCK2, PAK1, TBK1, TTBK1, IKKE, IMB1, IKBA, IL1, IL2, IL3, IL4, IL5, IL6, IL7, IL8, IL9, IL10, CD209) (Supplementary Table 1), were used as a basis to extract gene related expression data across samples for differential expression analysis. Their molecular functions, in relation to genetic interactions between host and virus as represented in the NCBI gene database (http://www.ncbi.nlm.nih.gov/gene), were considered across samples. Two other parameters considered were if the genes were upregulated and downregulated at least 2 fold in EBOV samples, which were extracted to complete the total list of genes (452 genes) used for expression comparative analysis across samples. The dominant genes observed on the EBOV interaction map, as selected above, were extracted separately into a gene expression heatmap with the aim of comparing how LCK gene, previously shown to be highly involved with immune functions (Achinko & Dormer, 2015), was related to other signaling genes, with special emphasis laid on the various MHC class I and II genes and CD209. LCK gene variants were compared to those seen for HLA and CD209 with the aim of understanding gene functional variation (Supplementary Table 1) and population immune variation related to the disease. The main MHC I HLA immune gene variants (A, B and C) most observed in the data were individually analyzed to see which immune variant occurred more often within the various samples.
For each gene, the RMEAN data was extracted and processed in two sets using a unique code (Achinko, 2016). One set considered the sum of all extracted values per gene per sample and then normalized across samples, while the other set was not normalized. Gene values per sample were entered into a table of values using a shell script. Data analysis for various statistical parameters were later processed using R version 3.3.1 (R Development Core Team, 2008; RRID, SCR_001905) (Dataset 1; Achinko et al., 2016a). For the normalized table, gene values were summed across samples (GS, gene summed) and all genes with GS = 0 were removed from the table and a minimal value of 0.001 was added to every value, so as to eliminate zeros from the table. The final normalized value per gene per sample was the Log2 [(Sample value + 0.001)/GS], and all values were considered for cluster analysis using Cluster version 2.0.4 with algorithm PAM wherein the input is a distance matrix from daisy (Kaufman & Rousseeuw, 1990) with metric=gower, a dissimilarity coefficient (Gower, 1971), and a cluster coefficient of 2. PAM is more robust; hence minimizing the sum of dissimilarities in values. The graphical representation from PAM clustering used the silhouette parameter (Rousseeuw, 1987) for interpretation and validation of clusters by showing which objects (genes) properly sit within their cluster or somewhere in between. The silhouette width is considered for clustering evaluation with the average used for validating clusters; hence used for selecting the number of clusters (e.g, two clusters in our case). Linear model analysis (Gelman & Hill, 2007) was used for t-statistics (R Core Team, 2013), which compared different parameters (skewedness and kurtosis) and F-Test statistics (R Core Team, 2013) was considered as a basis to accept or reject the null hypothesis of equal variance between samples at P value < 0.05. ANOVA two-way factorial (R Core Team, 2013) was used to compare pairwise expression patterns between samples from identified clusters. Using gplots version 2.1.0 (Gao et al., 2015) and RColorBrewer version 1.1–2 (Neuwirth, 2014), the data was subjected to hierarchical clustering (R Core Team, 2013; RRID: SCR_009154) and plotted using heatmap.2. Principal component analysis (PCA) performed on the data was done using ggbiplot library version 0.55 (Vu, 2011), which uses a covariance biplot scaled to one, wherein the covariance is approximated to the inner product between the variables while distances between points approximates to Mahalanobis distance (Mahalanobis, 1936).
Based on the query parameters, 54 samples were identified in GEO (Dataset 2; Achinko et al., 2016b), with genes ranging from 8,000 to 154,000, as submitted by original studies (Figure 1). These samples were further filtered for Homo sapiens only and 34 samples, including Zaire EBOV, formed the final list used for further analyses. The final list of 34 samples (Table 1) cut across different viral related diseases known to infect humans as host, with different pathogenic effects.
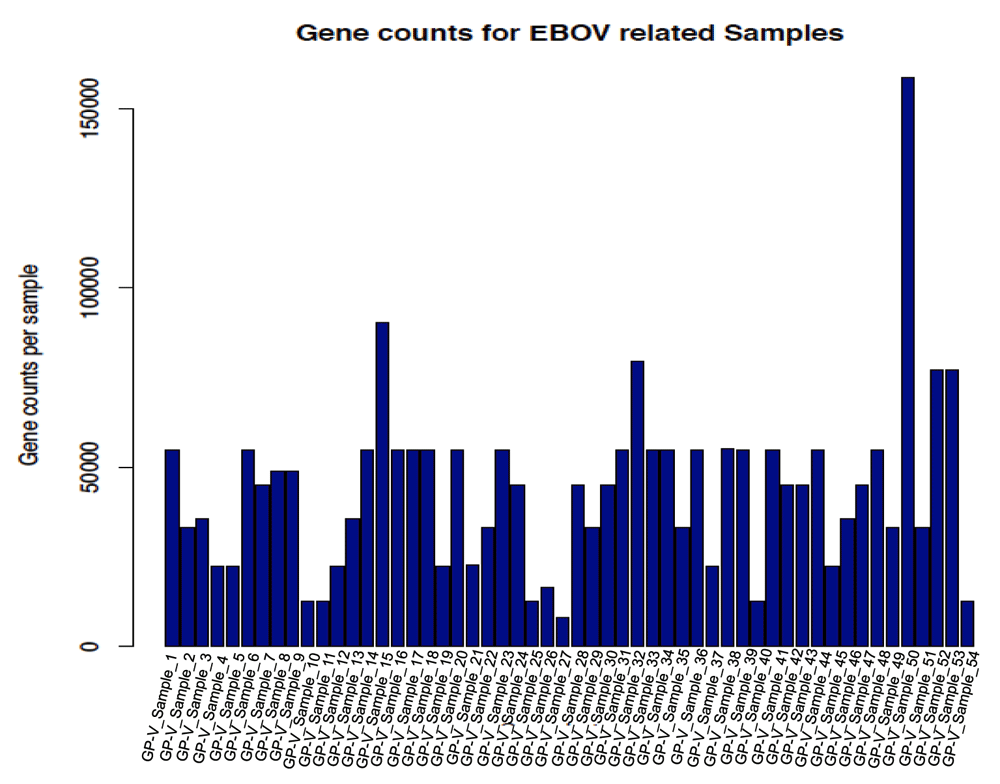
A total of 54 samples were identified for both human and mouse hosts. Genes expressed showed counts varying from 8,000 to 154,000 genes for different samples. EBOV, Ebola virus; GP-V_Sample_1, Glycoprotein and virus sample 1.
In total, 34 viral samples related to studies involving humans as hosts were obtained and the Relative mean (RMEAN) was further used to evaluate differential expression patterns across samples. The sample codes on the left was used for data graphs and the heatmap codes to the right were used for heat map figures.
From the collected data, normalization showed a relative range from 0 to 140 RMEAN frequency values (Figure 2). This large mean variation observed in some samples resulted in a linear model analysis of the mean in relation to skewedness and kurtosis of the dataset. The regression model showed an adjusted R-squared value of 0.3814, implying non-linearity. The median of residuals was equal to -0.01912, with many samples below the regression line and having negative values. The F-statistic value was 11.17 > F critical value of 2.1646, for 2 and 31 degrees of freedom. This was significant with P value = 0.0002219 and α=0.05, hence rejecting the null hypothesis and supporting strong differences within samples. The t statistics for the data showed significant values at α=0.05 (0.01496) for skewedness and α=0.01 (0.00814) for kurtosis, indicating significant differences in the respective parameters. Plotting the graph for residuals (y-axis) against fitted sample values in the model (x-axis) (Figure 3) showed that residual values ranged from -0.1 to +0.2, and though random, some few points deviated considerably from the regression line and this pattern was observed in three main samples [EB_Sample_1, EB_Sample_2 and EB_Sample_3 (Table 1)], which also clustered together. The log2 transformation of the data, as a means to observe relative gene expression patterns across samples, showed mean gene values per sample greater than zero, but with confidence intervals ranging from -5 to +5 (Figure 4). The total relative mean variation per sample showed no big difference between samples. This distribution of of the samples led to a random selection of the sample per cluster with the most gene related data, which were: sample 1 (EB_GEO_Sample_1) from the kurtosis graph cluster (KGC) (Figure 3), which also included samples 2 and 3; sample 27 (EB_GEO_Sample_28) from the large mean deviation cluster (MDC) (Figure 2), which also included samples 28 and 29; and sample 30 (EB_GEO_Sample_32) from the EBOV cluster (EBOVC), which also included samples 31, 32, 33 and 34. The selected samples were from studies concerning the following: sample 1, c-Myb and oncogenic variant v-Myb transcriptional activities; sample 27, Sindbis virus-induced cell death; sample 30, Zaire EBOV time course infection of macrophages. Samples 1 and 27 were each paired with sample 30 for comparative analysis. For gene count per sample versus log2 expression values, it was observed that samples 1 and 30 had a wide log2 expression coverage (Figure 5A), with the former showing more samples expressed with a fold change >2 for ≥ 30 genes. This distribution could account for the low values observed for skewedness and kurtosis in these samples. Comparatively, samples 27 and 30 showed a similar log2 expression coverage pattern, but the former had most of its gene expression concentrated between 0 to +2 fold change with very high gene counts (Figure 5B); hence possibly accounting for the high skewed and kurtosis values observed previously. The comparative analysis for each pair of samples representing the clusters related to gene expression patterns observed within clusters. Statistical analysis using ANOVA with a two-way factorial design showed a statistical difference for sample 30 (EBOV) compared to sample 27; F value = 191.415 and P value < 2e-16 and sample 30 compared sample 1; F value = 18.712 and P value = 1.88e-05. The difference between sample 1 and sample 27 was not significant (Figure 5C) with F value = 0.248 and P value = 0.619. Therefore, EBOVC showed significant differential expression patterns with KGC and MDC, but KGC and MDC showed no significant differential expression patterns.
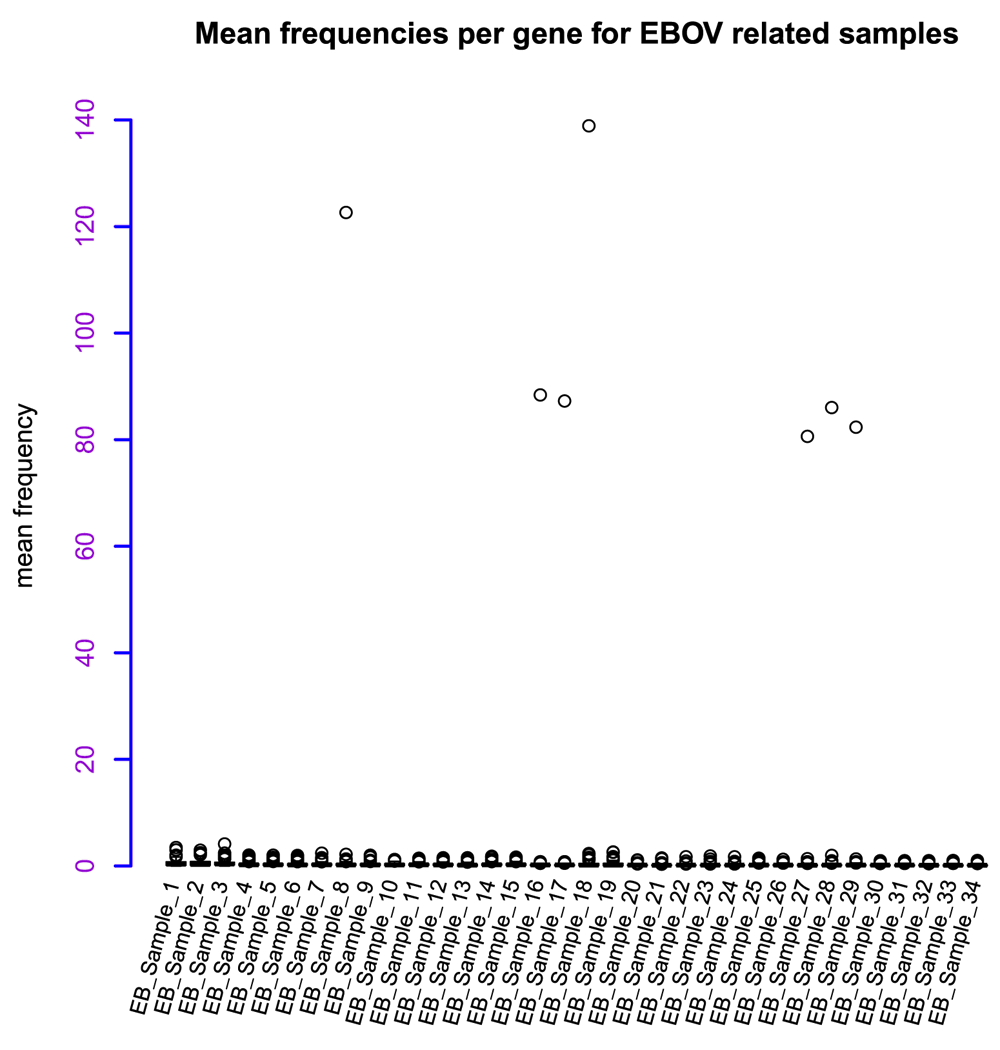
In total, 34 samples related to human hosts showed gene expression mean frequency variation ranging from 0 to 140. All of the samples had their mean close to 0, but with significant deviations seen in a few samples. EB_sample_32 was specific to EBOV. EB_sample, Ebola virus related sample; EBOV, Ebola virus.
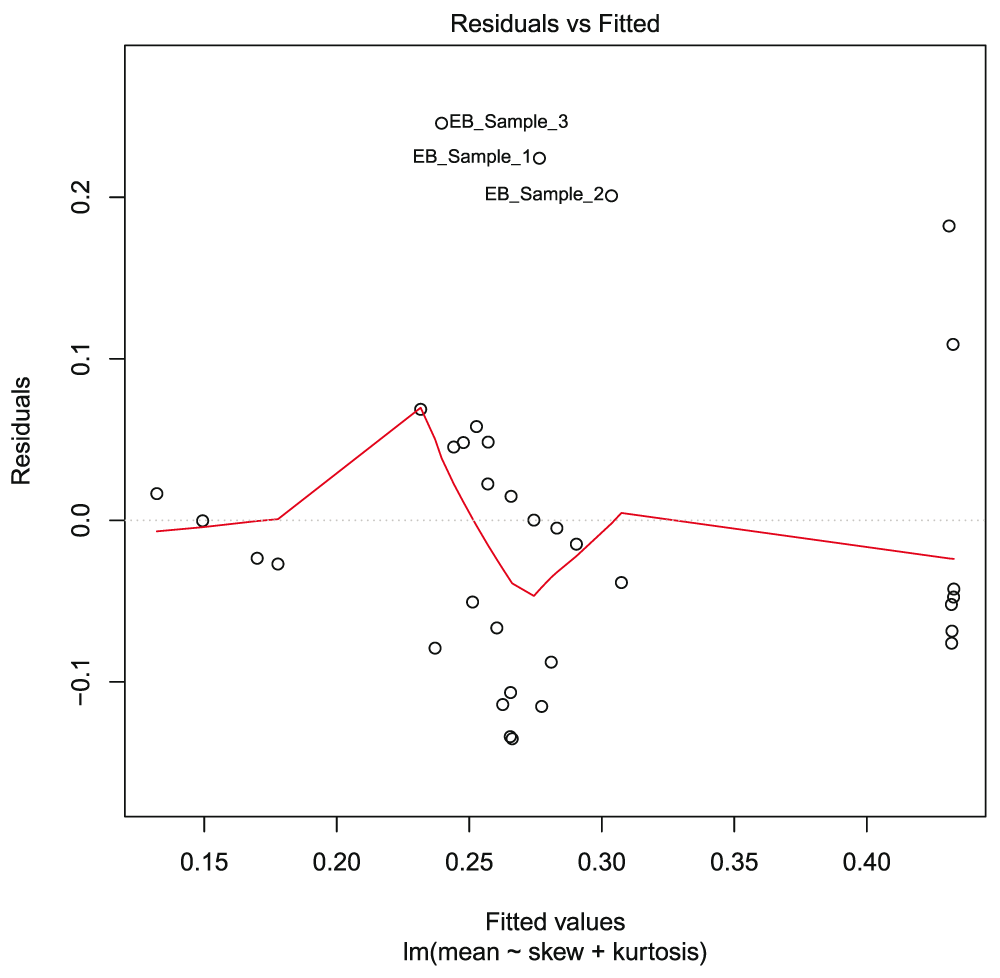
A linear regression analysis for mean against skewedness and kurtosis (mean ~ skew + kurtosis) showed that though the residuals showed a random distribution it wasn’t distributed properly for a straight line through the points. EB_Sample_1, EB_Sample_2 and EB_Sample_3 clustered together with residuals showing a higher positive value. The residual median value = -0.01912; hence, several points were negative. The regression statistics showed adjusted R2 = 0.3814, F statistic = 11.17 with P value = 0.0002219. T statistics showed P value = 0.01496 for skewedness and P value = 0.00814 for kurtosis. This showed that the variation in the data resulted more from kurtosis.
The log2 fold change ranged from -5 to +5, and in all the samples it was observed that the mean distribution was <0, suggesting that the majority of genes in all samples were upregulated. EB_sample_1, EB_sample_2 and EB_sample_3 were more upregulated than the rest of the samples and was considered the (kurtosis group cluster because it also clustered in the kurtosis graph (Figure 3). EB_sample_27, EB_sample_28 and EB_sample_29 were considered mean deviation cluster because of the large mean variation observed in these samples (Figure 2). EB_sample_30, EB_sample_31, EB_sample_32 EB_sample_33 and EB_sample_34 were considered (Ebola virus cluster because of the grouping with the Zaire Ebola sample (EB_GEO_Sample_32)).
Comparative gene expression profile between sample 1 (KGC) and sample 30 (EBOVC) with the former showing the majority of its genes (25 to 35) between +2 to +4 upregulation, and the latter having majority of its genes (15 to 20) between 0 to +2 upregulation. The difference was statistically significant with F statistics = 18.712 and P value = 1.88e-05. KGC, kurtosis group cluster; EBOV, Ebola virus; EBOVC, EBOV related cluster.
Comparative gene expression profile between sample 27 (MDC) and sample 30 (EBOVC) with the former showing the majority of its genes (20 to 40) between +1 to +2 upregulation. Both samples showed a statistical difference with F statistics = 191.415 and P value = < 2e-16. MDC, mean deviation cluster; EBOV, Ebola virus; EBOVC, EBOV related cluster.
Comparative gene expression profile between sample 1 (KGC) and sample 30 (MDC) showed no statistical significance between samples. F = 0.248 and P = 0.619. KGC, kurtosis group cluster; MDC, mean deviation cluster.
Cluster analysis using the PAM package on the 452 genes extracted for this study, identified two main clusters for the relative gene expression profile across samples (Figure 6A), and had the following gene distribution patterns: cluster 1 (401 genes) with average silhouette width = 0.53; and cluster 2 (51 genes) with average silhouette width = 0.16. The total average silhouette width = 0.49, implying very few genes showed a substantial variation across samples. The expression heatmap (Figure 6B) showed a similar gene distribution pattern for hierarchical clustering as observed with PAM analysis, resulting in grouping sample 30 (Zaire EBOV) with samples 13, 30, 31, 33, 34. Though sample 13 showed a similar RMEAN distribution pattern with those first mentioned in the EBOVC (Figure 2), emphasis remained on EBOVC (blue side color) as originally indicated, for further discussion. This cluster showed more genes with a relative expression ranging from 0 to ≤ +2 folds in upregulation, meanwhile a few genes were relatively downregulated up to 6 folds; hence correlating the pattern observed for the 51 genes in the PAM cluster. The cluster for samples 1, 2 and 3 (grey side color) observed in the regression graph also stood out on the heatmap, showing a similar and relatively high gene expression pattern across these samples. An extraction of the three clusters (KGC, MDC and EBOVC) into a heatmap (Figure 6C) showed that sample 30 had an expression pattern much closer to MDC than EBOVC The heatmap for the selected groups of genes (Figure 6D) dominantly involved with signaling (LCK-HLA heatmap) were plotted against all HLA gene variants identified across samples, and though EBOV still clustered in the same pattern with similar samples, sample 13 clustered with KGC not EBOVC, indicating that immune centric gene expression could show more segregation on related samples. An extracted HLA centric heatmap (Figure 6E) for the three clusters showed a similar pattern seen on the cluster specific heatmap observed for all the genes. Similarly, sample 30 clustered more with MDC than EBOVC. Based on EBOVC, differential gene expression pattern showed no expression (green color in heatmap) for some genes (MHCII: HLA-DPA2, HLA-DPB2, HLA-DQB2, HLA-DQA2; MHCI: HLA-H, HLA-L, HLA-F-AS1;IL1 receptor; and CD209), a ≥ -2 to ≤ +2 fold change (black color on heatmap) for other genes, including the main MHCI genes (HLA-A, HLA-B, HLA-C and HLA-E, HLA-F) MHCII HLA-DRB1, IL2 receptor alpha and SRC proto-oncogene non-receptor tyrosine kinase, and some genes showed fold changes of > 2, including MHCII HLA-DQB1, HLA-DPA1, HLA-DRB4, HLA-DRA, HLA-DOB, HLA-DPB1, HLA-DMA, HLA-DMB, HLA-DRB3, HLA-DRB4, HLA-DRB5, HLA-DRB6, IL2 and LCK. The most expressed genes on the cluster specific HLA centric heatmap were HLA-DRB2, a MHC class II gene, and MYB, a proto oncogene transcription factor), with an upregulation of ≥ +4. Relative to other clusters, gene variations observed for CD209, LCK, IL2 and MYB were further discussed given their role in immune activation.
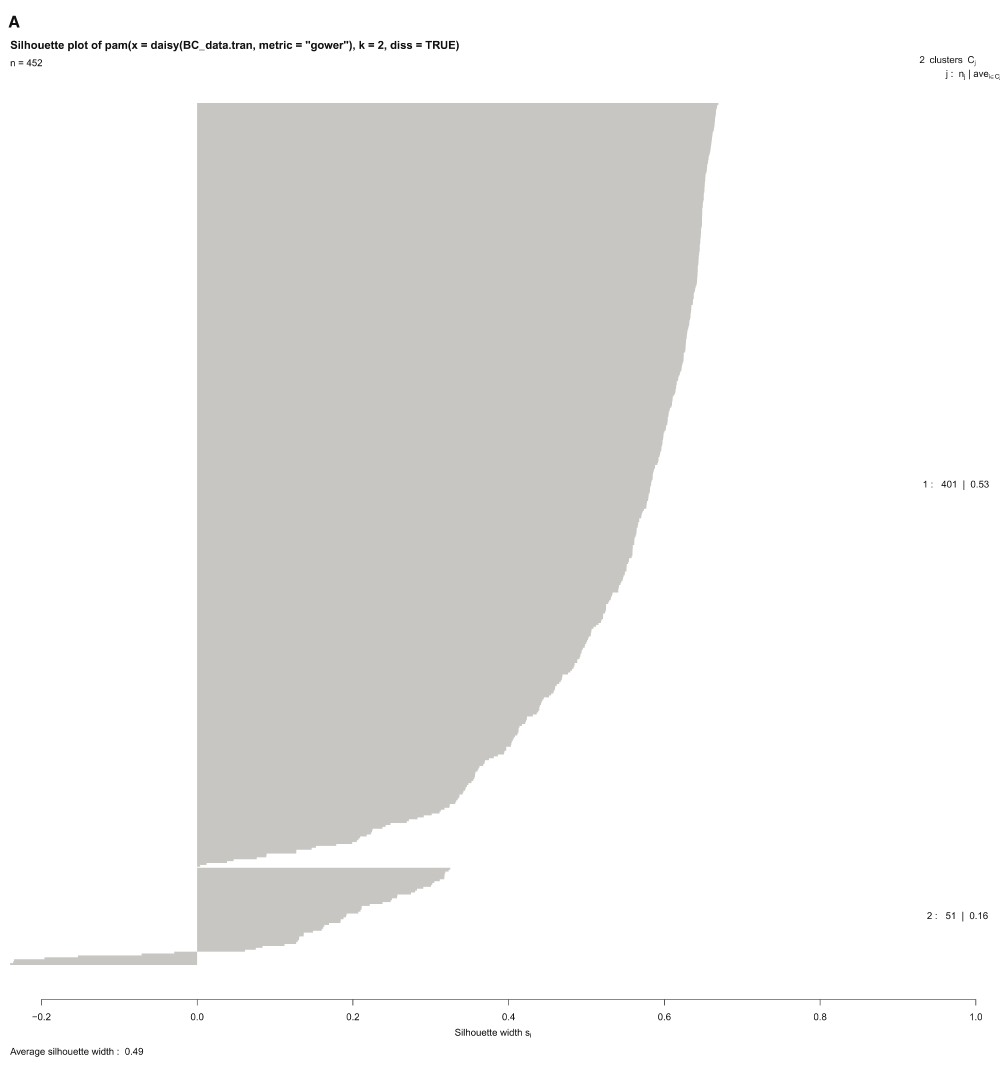
Silhouette cluster differentiation plot for all 452 genes used in the analysis. The PAM cluster package with metric gower was used to identify two clusters. The biggest cluster showed 401 genes and the second cluster showed 51 genes with some genes extending to the negative side of the silhouette width axis.

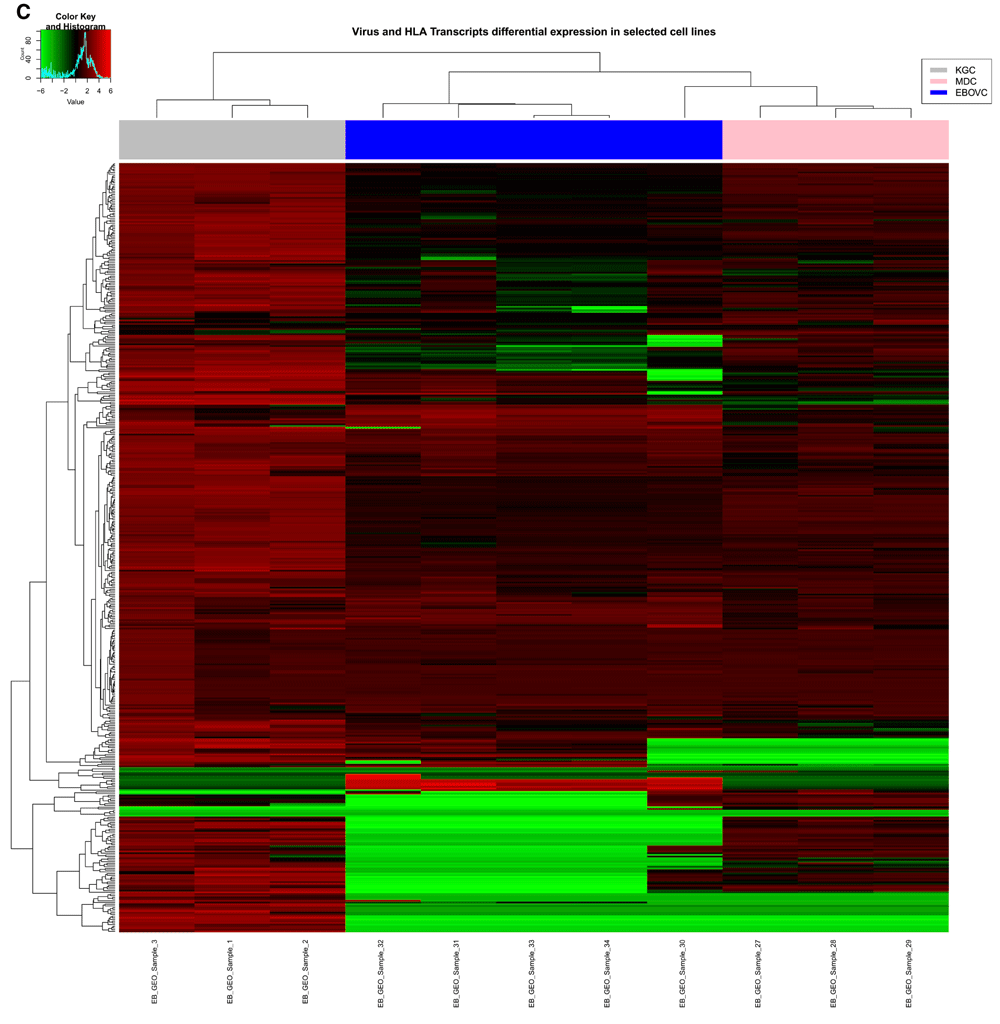
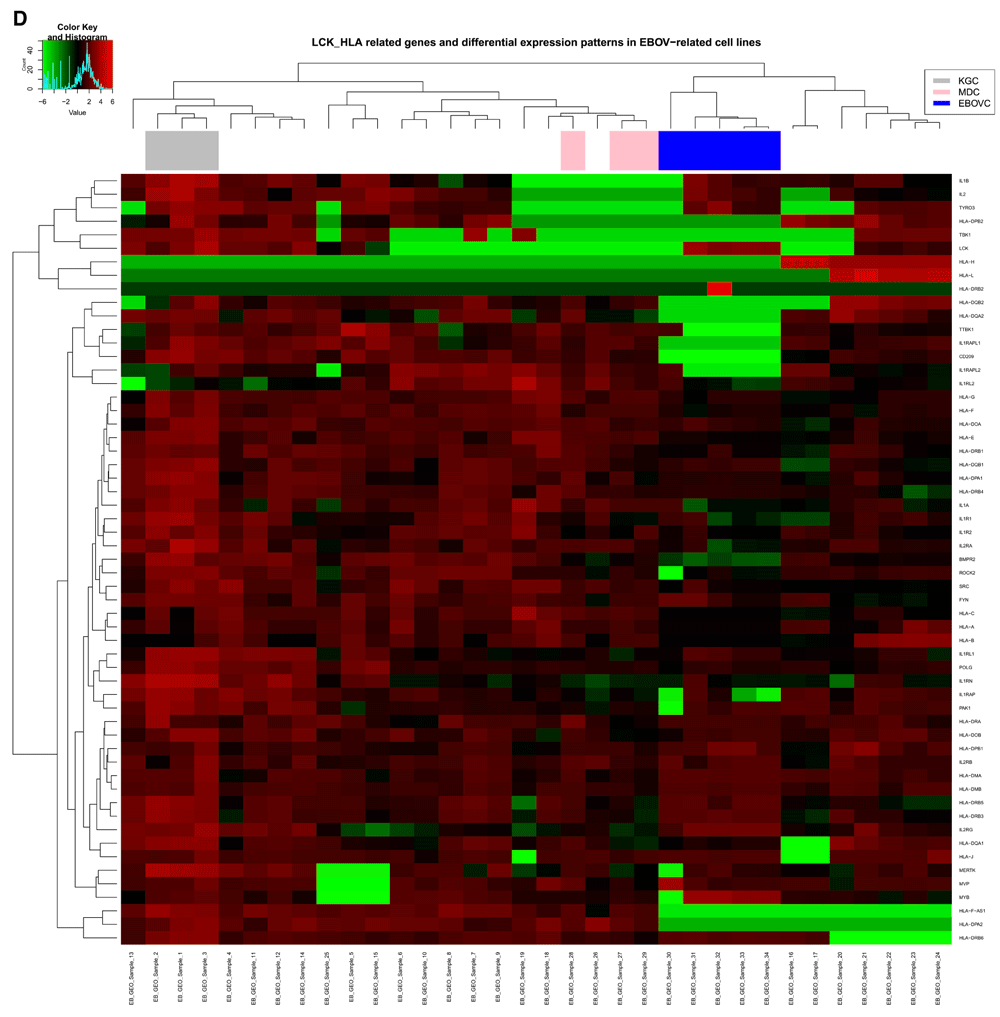
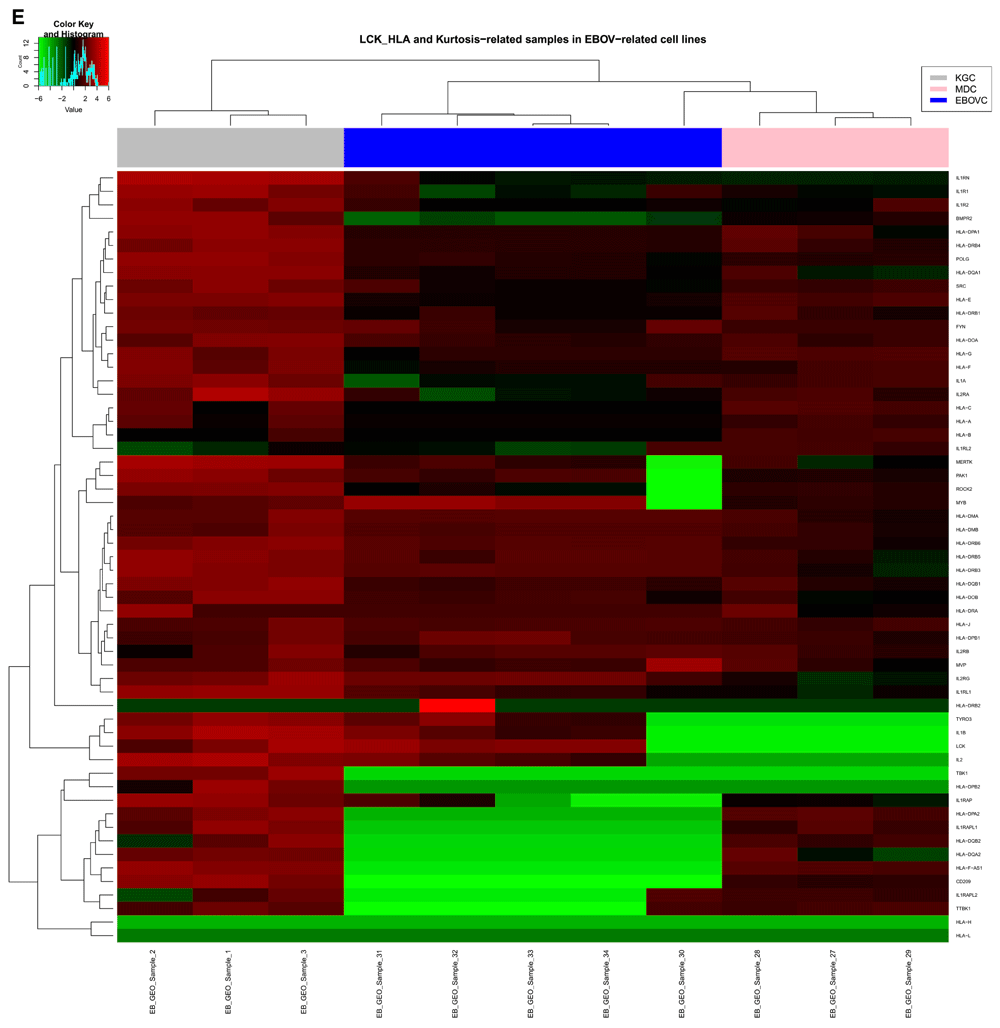
B) Heatmap for all 452 genes in this study with hierarchical clustering for genes on the y axis and all 34 samples on the x-axis. Samples in three main clusters related to KGC with 3 samples (EB_GEO_Sample_1, EB_GEO_Sample_2, EB_GEO_Sample_3), MDC with 3 samples (EB_GEO_Sample_27, EB_GEO_Sample_28, EB_GEO_Sample_29) and EBOVC with 5 samples (EB_GEO_Sample_30, EB_GEO_Sample_31, EB_GEO_Sample_32, EB_GEO_Sample_33, EB_GEO_Sample_34). These were highlighted with the following color codes for further analysis: KGC, gray); MDC, pink; and EBOVC, blue. EB_GEO_Sample_32 was specific to Zaire EBOV. KGC stood out with high expression profiles seen across its genes compared to all other samples. C) Heatmap for 452 genes specific to clusters of interest. The color codes define clusters of interest and EB_GEO_Sample_30 showed that it was closer to MDC than EBOVC, as originally observed. D) Heatmap of 57 genes with functions related to signaling and immune response. Genes were extracted from the total data, but all samples were considered for clustering pattern comparative analysis with total gene heatmap. Clustering pattern showed a variation on samples with specific observation for EB_GEO_Sample_13, which originally clustered with EBOVC, but now clustered with KGC. Color code represents clusters of interest. E) Heatmap for 57 genes specific to clusters of interest and showing a similar pattern to previous heatmaps for clusters and all genes. Genes of interest were CD209 (DC-SIGN), a C-type lectin expressed in KGC and MDC, but absent in EBOVC, and LCK, a lymphocyte specific tyrosine kinase, which is expressed in KGC and EBOVC, but not MDC. KGC, kurtosis group cluster; MDC, mean deviation cluster; EBOV, Ebola virus; EBOVC, EBOV related cluster.
The frequency distribution of all MHCI gene variants identified across samples were evaluated. It was observed that relative to HLA-B, which had the highest frequency distribution (100%), HLA-C had a 85% frequency distribution and HLA-A was 38%, while the top three MHCII genes ranked as follows: HLA-DRB1, 25%; HLA-DQB1, 16%; and HLA-DPB1, 15% (Figure 7). The three top MHCI gene variants considered also showed different frequency patterns across samples, with the following top 5 alleles: HLA-B*2704, HLA-A*2711, HLA-B*2702, HLA-B*1537 and HLA-B27052. The only HLA class I allele observed specifically for EBOV was HLA-B*2702. The top three MHCII gene variants considered belonged to: HLA-DPB1 with HLA-DPB1*0202, HLA-DPB1*0501, and HLA-DPB1*1301 alleles (accession number: NM_002121) and HLA-DQB1 and HLA-DRB1 with HLA-DR2-Dw12, DRw6, DQw1, and DQw9 alleles (accession number: NM_002123). The MHCII HLA variant alleles specific for EBOLA were HLA-DR2-Dw12, DR7, DQw9, DR2.3 and DQw2.
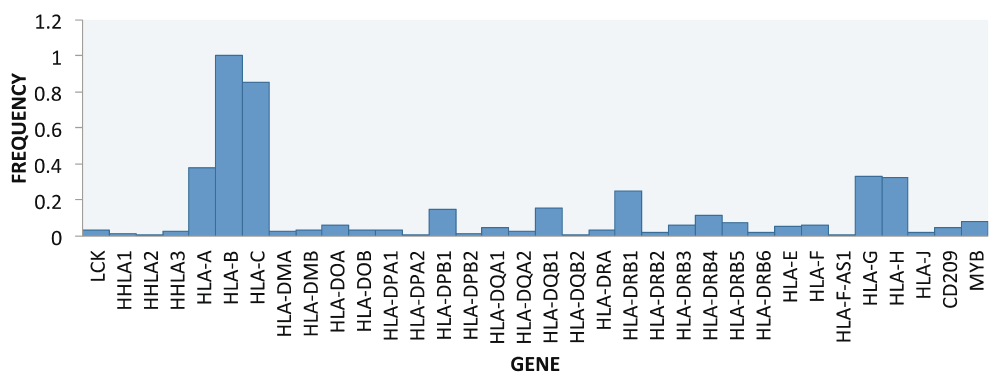
Immune genes for MHC HLA class I & II were extracted from the total data and to it three more genes of interest CD209 (DC-SIGN) a C-type lectin, LCK and MYB, a transcriptional activator, were added to the total data. HLA class I genes had the highest frequency: HLA-B was the highest (100%), followed by HLA-C (85%) and HLA-A (38%). For HLA class II genes, the highest was HLA-DRB1 (25%), then HLA-DQB1 (16%) and HLA-DPB1 (15%). LCK, lymphocyte specific tyrosine kinase; DC-SIGN, dendritic cell specific C-type lectin intercellular adhesion molecule 3 grabbing nonintegrin.
LCK is a known proto-oncogene of the Src family tyrosine kinase and is an important molecule involved in signaling, which relates to the selection and maturation of T-cells under development (http://www.ncbi.nlm.nih.gov/gene/3932). EBOV with related viral diseases, like HIV, are known to interact through their envelope GP molecule, with CD209 on the surface of dendritic cells to penetrate the host cell (Lin et al., 2003). However, it is not clear if LCK and CD209 do interact directly, but it has been shown that C-type lectins possessing the intracellular signaling immunoreceptor tyrosine-based activation motif (ITAM) interacts via SH2 domain on SRC kinases (Sancho & Reis e Sousa, 2012), and LCK have a similar domain. Based on our previous publication on EBOV antigenic peptides favoring successful viral transmission (Achinko & Dormer, 2015), LCK was identified as the main host signaling protein involved with immune related functions. The three clusters of interest, KGC, MDC, and EBOVC, showed three distinct patterns on the heatmap (Figure 6E) for LCK and CD209 gene expression. MDC showed a positive expression for CD209 [NM_021155 (Hijazi et al., 2011; Rappocciolo et al., 2006) and AF290886 (Bashirova et al., 2001; van Vliet et al., 2009)], but not LCK. EBOVC showed a positive expression for LCK [M26692 (Takadera et al., 1989), M36881 (Lesley et al., 2002; Wu et al., 1995) and U23852 (Vogel et al., 1995)], but not CD209. KGC showed expression for both LCK [NM_005356 and U07236 (Wright et al., 1994)] and CD209 [AF290886 (Bashirova et al., 2001; van Vliet et al., 2009), AY042224 and NM_021155 (variant 1: Hijazi et al., 2011; Rappocciolo et al., 2006)]. The differential expression pattern observed for IL2 and MYB genes was also considered because the former is known to be induced by LCK as its effector, leading to T-lymphocytes division (Vogel et al., 1995), while the latter is known to be an IL2 responsive gene and so becomes activated downstream of IL2 (Beadling & Smith, 2002). IL2 was expressed only in the KGC samples [NM_000586 (James et al., 2002; Lupino et al., 2010) and M22005 (Weir et al., 1988)] and EBOVC related samples [M22005 (Weir et al., 1988), S82692 (Chernicky et al., 1996) and X00695 (Holbrook et al., 1984)], while MYB was expressed in all three clusters, but the highest expression (> +2 fold up regulation) was seen in EBOVC. MYB variants for the various clusters were EBOVC [M13666 (Slamon et al., 1986), M15024 (Dash et al., 1996) and U22376 (Beadling & Smith, 2002)], KGC [NM_005375 (variant 2: Lorenzo et al., 2011) and AI357042 (V-myb myeloblastosis viral oncogene homolog (avian))] and MDC [NM_005375 (variant 2: Lorenzo et al., 2011) and AI357042 (V-myb myeloblastosis viral oncogene homolog (avian))].
For the PCA, the covariance biplot at scale = 1, generated scattered plots for the two main components considered, and data points were plotted within a correlation circle of radius = 1, based on the Mahalanobis distance, which looks at point variations away from the mean with a maximum deviation of 1 as belonging to the dataset or >1 as not belonging to the related dataset. Focusing on the total dataset, it was observed on the y-axis (PC2) that gene related variation explained 19.2% variation among samples, while on the x-axis (PC1) sample related variation explained a 22.8% variation across genes (Figure 8A). For the HLA immune centric genes, the y-axis (PC2) explained a gene related variation of 18.0% variation among samples while on the x-axis (PC1) sample related variation explained a 31.3% variation across genes (Figure 8B). Several genes were localized out of the correlation circle in the HLA immune genes centric group, including X-linked IL1 receptor accessory protein-like 2, IL1 receptor-like 2, IL1 beta (IL1B), tyrosine-protein kinase receptor (TYRO3), serine/threonine-protein kinase (TBK1), LCK, HLA-DRB2, HLA-H and HLA-L.
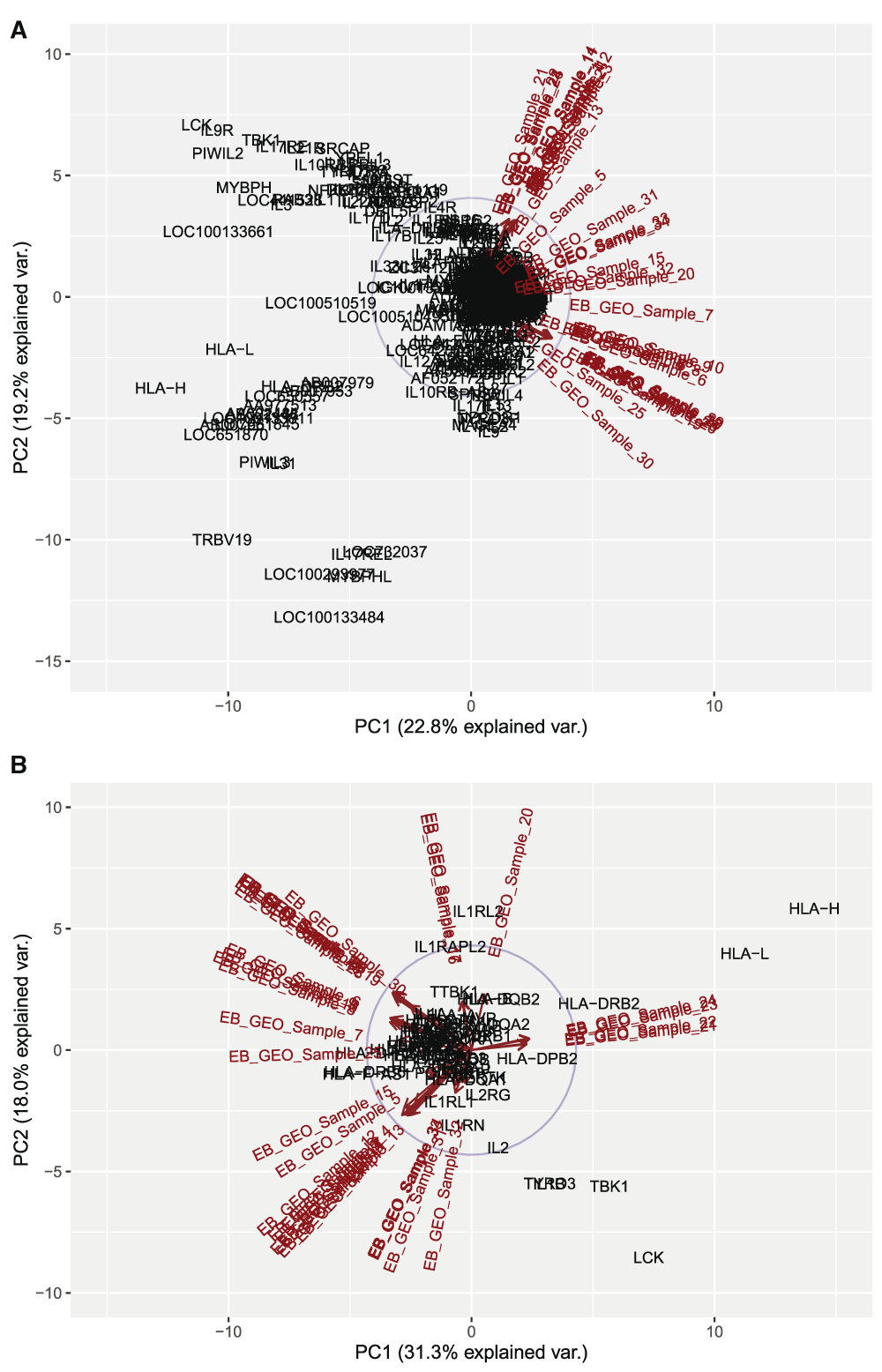
This is a covariance biplot analyses, which looks at the variance across genes and samples and plots them as a correlation circle with radius = 1, wherein a maximum deviation from the center and out of the circle considers the point of interest as not belonging to that dataset. A) Biplot for all 452 genes in the dataset and all 34 samples. In total, 19.2% variation could be explained across samples, while 22.8% variation could be explained across genes. This high variation in genes compared to samples could be seen by the grouping of samples on the plot compared to a more scattered pattern for genes, and with LCK, one of the genes of interest seen out of the plot. B) Biplot for 57 genes with signaling and immune response functions across all 34 samples. In total, 18.0% variation could be explained across samples, while 31.3% variation was seen across genes. The gene related variation placed LCK out of the correlation circle as not part of the dataset.
The concept of differential gene expression has always been referred to datasets where, for a particular diseased condition of interest, data is collected for the diseased state (experimental) and the non-diseased state (control), and the difference in gene profiles between the two conditions helps explain the gene regulation pattern within the cell at a given time. This becomes a challenge when post analysis of the data is very difficult to address if no comparative and related well-studied dataset is available; hence, the data ends up with more questions to be answered than solutions to address the diseased condition. This has been the case when addressing the EBOV disease burden in 2014, which lead to the death of numerous people and left many families in pain (Chiappelli et al., 2015). Therefore, there is a need to exploit the currently documented EBOV disease data alongside virus related data with similar genetic underpinnings to better understand the common and related disease pathways that could be further molecularly characterized to better arrest EBOV in the future.
The viral GP envelope remains the main genetic component identified by CD209 on human DCs, as a target for generating an immune response (Achinko & Dormer, 2015). For the 34 human related virus data samples obtained for analysis based on our search terms, the gene related data ranged from 8,000 to 154,000 genes for different samples. This genetic profile depicts a viral related expression variation pattern in a wide majority of genes within the host, and a large number of them co-regulated in a similar fashion across different viral diseases, as seen in the collected samples. Hence, genetic comparative analysis of gene expression profiles between related samples could tell a better story on immune regulatory patterns and viral targets, which could be exploited for disease prevention. Regression analysis of the mean against skewedness and kurtosis showed a non-linear pattern across residual points, though a random pattern was observed in the residuals. The statistical analysis with P value < 0.05 confirmed variation in the samples with more significance for kurtosis than skewedness. Kurtosis, which is a measure of the heaviness of tails in a data, considers higher kurtosis values as extreme and less frequent deviations of the variance in the data, compared to frequently and modestly sized deviations (Westfall, 2014). Therefore, MDC with high kurtosis values suggests that gene expression distribution is not uniform across each sample, with some genes showing extremely high and low expression patterns (Figure 5B). Samples in MDC showed variable genetic expression patterns related to genes of interest that is, up regulation of CD209 and MYB and down regulation of LCK and IL2. This cluster showed similar genes of interest expressed with KGC and not EBOVC. Virus samples in this cluster showed an inhibition of IFNs, which are activated downstream of Pattern Recognition Receptors (PRRs) like Toll-like receptors (TLRs) on epithelial and dendritic cells. Proinflammatory cytokines are also activated through this pathway, therefore the absence of LCK suggest that this gene could be activated downstream of this pathway and induction of IL2, a non-inflammatory cytokine, by LCK, further suggest a downstream pathway specific to activation of immune T-cells. Sample 27 (Laine et al., 2004) showed that Sindbis virus is an arthritis related arbovirus of the alpha virus group and is transmitted by insect vectors. Arboviruses and likewise EBOV, commonly cause disease outbreaks, resulting in high fatality rates. Sindbis virus is able to infect neurons, hence decreasing immune responses associated with type I and II IFNs (Griffin, 2010) through the Janus kinase (JAK)/STAT pathway (Simmons et al., 2010). The observed expression pattern in this sample was similar to sample 29; Bronchial epithelial cell line response to various airway pathogens. An example of airway pathogen with this pattern was Parainfluenza Virus 3 shown to block downstream antiviral mediators like IFN by modulating STAT1 phosphorylation (Eberle et al., 2015). Sample 28 (Schlee et al., 2004) showed that Epstein-Barr Virus (EBV) is a B-lymphotropic herpes virus and post infection of B-cells, nuclear antigen 2 (EBNA2) is one of the first viral genes expressed. EBNA2 is a transcriptional activator regulating expression of viral and cellular genes, which initiate and maintain cell proliferation. EBNA2 is needed for virus infected cells proliferation, establishing viral latency in large numbers of B-cells (Babcock et al., 2000). The strong transactivation domain of EBNA2 does not interact with DNA directly but rather with several components of RNA polymerase II transcription complex (Tong et al., 1995). Through sequence specific DNA, EBNA2 gets recruited to the promoter of target genes and can upregulate genes like CD23 and CD209. MDC showed that the inhibition of the IFN pathway in these related viruses leads to B-cells infection and possible evasion of MHC class I pathway shown to occur through EBNA1. The interaction of EBNA2 with c-myc confirms the upregulation of CD209 in these viral samples. Dendritic cells are professionally involved in antigen presentation with immune related function. Upon antigen capture from the periphery, their activation results in migration to lymphoid tissues and presentation of antigens to T-lymphocytes creating a pathogen-specific immune response (Janeway et al., 2004). C-type lectins, on DCs are known to possess carbohydrate recognition domains for interaction with GPs on pathogens, but some lectins possess a C-type lectin-like domain, which is non-specific to carbohydrate binding and is found on C-type lectin-like receptors (Geijtenbeek et al., 2004). This is the case of CD209 belonging to the DC-SIGN lectin group. CD209 lacks the ITAM required for LCK interaction and activation for immune induction (Sancho & Reis e Sousa, 2012), but it presents with a non-immunoreceptor tyrosine-based motif and is specific to DCs (Geijtenbeek et al., 2000). CD209 contains two dileucine internalization motifs and is known to mediate interactions between DCs and resting T-cells through the interaction with intercellular adhesion molecules (ICAM-2 and 3); however, the use of antibodies against DC-SIGN inhibits the induction of T-cells by DCs by >60%, (Geijtenbeek et al., 2000), suggesting that DC-SIGN (CD209) acts as an immune escape route. Through receptor mediated antigen endocytosis on DCs, antigens are processed by the endosomal or lysosomal pathway to form MHC class II peptide complexes for further presentation to CD4+ T-cells (Pieters, 2000). From our data, MHC HLA class II peptides were highly expressed in all clusters, with HLA-DRB2 being specific to EBOV. The ITAM domain interacts with the LCK gene through SH2 domains, and the low expression profiles observed for MHC HLA class I genes in the MDC cluster suggest that the lack of interaction between CD209 and LCK is critical for MHCI activation and subsequently, CD8+ T-cell induction. Therefore, DC-SIGN should induce MHC HLA class II genes through ICAM motifs, while other DCs with the ITAM motif, which interact with LCK, should induce MHC HLA class I genes. Therefore, other possible DC routes require urgent identification for antiviral therapy and immunotherapy needs to take into consideration the inhibition of CD209 route to completely eliminate viral infection.
For KGC, the kurtosis values were not high, but their pattern on the graph (Figure 3) showed considerable deviation of residuals from the regression line with a high positive value. This pattern was consistent with upregulation of almost all genes in these samples, which were much higher than those observed for EBOVC, as seen in Figure 5A. This observed pattern could suggest that strongly positive samples with high residual deviation on a regression plot for the mean against skewedness and kurtosis present with >90% of genes upregulated, including immune related genes (HLA class I and II). This cluster showed a statistical difference with EBOVC and not MDC suggesting similar expression patterns with the latter and not the former. KGC showed similar CD209 and MYB genes with MDC while with EBOVC, it showed different LCK and MYB genes but similar IL2 genes. Sample 1 (Quintana et al., 2011) showed that c-Myb transcription factor is involved in hematopoietic proliferation and its inhibition causes lethal anemia and lack of T- and B-cell development in mouse embryos. v-Myb is a transcriptional activator and is also considered an oncogene transduced by retroviruses known as avian leukemia virus E26 (ALV) and avian myeblastosis virus (AMV), due to truncation and mutations (Lipsick & Wang, 1999) at its C-terminal, also known as the 3’ end. The C-terminal of c-Myb presents four domain types conserved in chicken, mice and humans, which include transcriptional activation domain (Dubendorff et al., 1992), FAETL, an oncogenic activity domain, (Fu & Lipsick, 1996), TPTPF, which is conserved in MYB genes, and EVES (negative regulation and molecular interaction) (Dash et al., 1996). Mim-1 was the first chicken gene identified to be regulated by c-Myb and E26 v-Myb, but not AMV v-myb. The three consecutive binding sites at the Mim-1 promoter region are known to interact with v-Myb (Ness et al., 1989), and both c-Myb and v-Myb activate their genes through the PyAACG/TG motif (Biedenkapp et al., 1988). It has been shown that enhanced alternative RNA splicing in leukemia samples can result in ~60 different mRNA variants encoding at least 20 different c-Myb versions, since at least 6 alternative exons and several splice donors and acceptors do occur in its standard exons. The many c-Myb variants show differences only at the C-terminal site, and this leads them to differentially target genes based on selected promoter interactions (O’Rourke & Ness, 2008); hence asserting the fact that enhanced splicing in leukemias produce oncogenic and truncated c-Myb variants contributing to leukemogenesis. Sample 2 (Sirois et al., 2011) showed that HIV-1 also infects macrophages, using it as potent reservoir to rapidly disseminate throughout the body and also infect CD4+ T-cells through a viral synapse. HIV-1 infection on macrophages induces IFNs and Interferon stimulating genes (ISGs) through innate immune response, which further stimulates a polypeptide like-A3 of the apolipoprotein-B mRNA-editing, enzyme-catalytic (A3A) family known to be critical for monocyte resistance to infection. Decreased A3A expression during differentiation of macrophages leads to an HIV-1 vulnerable target cell population (Peng et al., 2006) hence circumventing the IFN protective effect in these cells. Sample 3 (Lung et al., 2014) showed that EBV is a major player in the development of nasopharyngeal carcinoma, which is an epithelial cancer. As previously seen above for sample 28, EBV through EBNA2, preferentially infects B-cells and other immune cells leading to tumorigenesis, which could actively occur through its direct activation of c-myc oncogene (Kaiser et al., 1999). KGC showed a dominant regulatory pattern of genes through transcription binding and mRNA spliced variants. It demonstrates a genomic pattern in cells similar to tumors, and is mainly caused by oncogenic variants of transcription factors involving two avian virus types (ALV and AMV). The C-terminal mutations associated with v-Myb and c-Myb have been implicated in oncogenesis, and given that c-Myb C-terminal mutations are involved with leukemogenesis, chronic lymphocytic leukemia also shows wide intron retention resulting from mutations on the spliceosome factor SF3B1, incapacitating it to properly splice 3’ ends of nascent mRNAs (Wan & Wu, 2013). In this cluster we also observed evasion of the immune system and direct infection of immune cells. The transcription directed role of c-myb and v-myb suggests an active involvement and specificity of these proteins in immune cells leading to leukemogenesis.
In the EBOV cluster, Sample 31 (Zilliox et al., 2006) demonstrated that measles virus is associated with mortality and resultant life long protection. It has been shown that measles virus can infect monocytes derived immature dendritic cells and replication occurs through dendritic cell maturation and subsequent activation of T-cells (Servet-Delprat et al., 2000). Infection of dendritic cells by measles virus leads to down-regulation of some cells (CD40-CD40L signaling co stimulating TCR/CD3, IL-12 and CD4+ T-cells) and upregulation of TNF-related apoptosis-inducing ligand to initiate apoptosis in T-cells. Sample 32 (An et al., 2005) showed that Kaposi’s-sarcoma associated herpesvirus could be the main cause of Kaposi sarcoma, primary effusion lymphoma (lympho proliferative disease) and Castleman’s disease. Most cells with these malignancies are latently infected, expressing in majority latency-associated nuclear antigen (LANA) a viral latent gene (Dupin et al., 1999). LANA is a multifunctional protein and functionally related to EBNA-1 of the EBV earlier discussed (Sample 28), which was seen to escape MHC class I pathway. Sample 33 & 34 (Rieger et al., 2004) showed that radiation toxicity on EBV immortalized B-cells were in majority associated with DNA damage leading to abnormal transcriptional responses. The virus distribution pattern in EBOVC showed that viral infections affect transcriptional responses in infected cells leading to DNA damage. The pattern in dendritic cells showed a TNF upregulated apoptosis of T-cell induced pathway suggesting massive destruction of cells. The upregulation of dysfunctional LCK in this cluster suggests a viral transcriptional regulatory pattern in infected cells possibly resulting from related genes like LANA and EBNA1 and EBNA2, leading to several LCK variants expressed in DCs. Downregulation of CD209 in this cluster shows that some viruses possibly use an alternate route through dendritic cells and for measles viruses, it has been shown that, CD150 (De Witte et al., 2008) a signaling lymphocytic activation molecule [SLAM (http://www.uniprot.org/uniprot/Q13291)] involved in the interaction of both innate and adaptive immune systems, is most used. SLAM is mainly expressed on thymocytes, lymphocytes, macrophages and mature DCs (Yanagi et al., 2006). SLAMF1, the T and B lymphocytes SLAM version was shown to recruit FYN (Tyrosine protein kinase) through its cytoplasmic adapters for its phosphorylation and FYN was seen clustering with LCK in our previous work (Achinko & Dormer, 2015). SLAMF1 also mediates proliferation of activated T-lymphocytes by IL-2 independently during an immune response (Aversa et al., 1997) and this IL2 related activation has been shown previously for LCK (Vogel & Fujita, 1995). For innate immune responses, SLAM recruits PI3K complex for response against Gram-negative bacteria in macrophages (Berger et al., 2010) and PI3K also acts downstream of LCK. SLAM could be a possible route used by EBOV through its soluble glycoprotein molecule to penetrate the cell unlike the structural glycoprotein used by various viruses. Differentially mutated LCK variants were expressed in KGC for related viral infected immune cells and EBOVC for dendritic infected cells, suggesting that, the dual role played by SLAM could engage LCK protein in different functional pathways due to its differential regulatory pattern in lymphocytes, dendritic cells and macrophages. Herpesviruses associated with EBOVC has been shown to encode proteins having inhibition effect on MHC class I antigen presentation molecules, hence helping the virus evade cytotoxic T-cells. MHC class I molecules are located in the endoplasmic reticulum (ER) where they fold, assemble and bind 8–10 amino acids of antigenic peptides, all required for the assembly process. Therefore viral peptides need to be translocated to the ER and this process is facilitated by the transporter associated with antigen processing (TAP) protein (van Endert et al., 2002), which further presents a scaffold for binding while ER chaperones fold novel MHC class I molecules, respectively. Tapasin protein forms the critical link facilitating the binding of pathogen derived peptides and MHC class I derived variants to TAP, after which, the MHCI-peptide complex are selectively transported through cargo vesicles to the Golgi apparatus (Spiliotis et al., 2000). The herpes simplex virus ICP47 cytosolic protein exploits the TAP role in MHCI-peptide assembly by acting as a competitive inhibitor of peptide binding (Neumann et al., 1997) hence preventing the MHCI-peptide formation complex. The MHC class I molecules were most downregulated in EBOVC compared to other clusters but in general the expression in all clusters was < 2 folds. The effect of herpes proteins had functional similarities with EBV whose protein effects showed different regulatory effects through EBNA1, EBNA2 and c-myc in the viral infected cells. The differential regulatory pattern of LCK in dendritic cells and immune cells suggests an interaction pathway which could be key to TAP and MHCI proper functioning and exploited by several viruses for their pathogenic gains. Therefore proper understanding of viral inhibition pathway and LCK interaction could help in developing therapies favoring immune activation against viruses through the MHC class I molecules. Previously seen in Sample 27 (MDC) that inhibition of the IFN pathway could result in LCK downregulation, suggest a possible activation route for LCK whose upregulation in this cluster resulted downstream of IFNs and cytokines production after DC activation. During an infection (viral or bacterial), innate immune response results in activation of PRRs, such as Toll-like receptors and CD209 on DCs, histocytes, Kupffer cells and macrophages, with subsequent release of inflammatory mediators (histamine, bradykinin, serotonin, leukotrienes and prostaglandins), which produce pain sensation and vasodilation of local blood vessels; hence attracting neutrophils as the main phagocytes to the affected site (Stvrtinová et al., 1995). This phenomenon should be the cause of exacerbation of gene expression and disease shock syndrome, commonly known as the cytokine storm (Mackenzie & Lever, 2007), seen in those infected by EBOV in the 2014 epidemic. Therefore downregulation of LCK could lead to viral inhibition of MHCI in infected cells permitting these cells to directly infect CD4+ T-cells when they migrate to lymphoid tissues.
LCK is a T-cell specific leukemia related gene, found in several cancer cell types, possessing two promoter regions whose differential usage leads to LCK type I mRNA for proximal promoters and type II mRNA for distal promoters. Type II mRNAs have been shown to be dominantly expressed in T-cell leukemia and two colon cancer cell lines (Takadera et al., 1989). It was observed that type IIB mRNA was void of exon 1, which encodes LCK N terminal domain; hence lacking the sequence coding motif required for interaction with CD4 and CD8 co-receptors (Huse et al., 1998). Alternative splicing at the distal promoter with many splice sites produces another mRNA lacking exon 7, retaining intron B, which lacks the ATP binding site and is also very unstable (Nervi et al., 2005). This mRNA, void of exon 7, shows a deficient signaling pattern downstream of the complex CD3/TCR in an early stage immunodeficient patient (Goldman et al., 1998). LCK has also been documented to regulate CD28, one of the most efficient receptors required for T-cell activation and also PI3K (Gibson et al., 1996). IL2 has been shown to induce human peripheral T-cell lymphocytes to re-enter the maturation phase from rest (G0) (Meyerson & Harlow, 1994). It is activated through a receptor associated with catalytic activity by LCK. c-Myb is known to lie downstream of IL2 (Lauder et al., 2001); therefore, LCK interaction pathway could involve SLAM, TAP, PI3K, IL2 and c-Myb whose differential interaction in related cells drives gene transcription related to T-cell activity. This is the first time a suggested immune pathway relative to T-cell immune induction involving LCK required to arrest viral pathogenesis has been identified. Further investigation of this information could be key to preventing viral disease burden.
The CD209 variant in KGC showed that upon interaction with HIV-1, T-cell infection and pathogenesis by the virus is enhanced (Bashirova et al., 2001). Although it’s not clear if there is any direct genetic interaction between CD209 and LCK, CD209 and its signaling complex involves lymphocyte specific protein 1 (LSP1) and Raf-1, a serine/threonine-protein kinase, which upon activation results in a T-cell infection common to HIV (Gringhuis et al., 2009). The LSP1 gene suggests a similar functional class for LCK, which is also lymphocyte specific. In a general sense, viruses tend to use the DC-SIGN route to escape lysosomal degradation and thereby deviate from immune surveillance (Ludwig et al., 2004), leading to CD4 T-cell infection. Therefore, infection of CD4+ helper T-cells, common to HIV and KGC, suggest a pathway used by HIV and related viruses like EBV to escape immunity clearance by the cell. The statistical difference between clusters, as depicted by sample comparative analysis (Figure 5A and B), was statistically significant for EBOVC compared to KGC and MDC, suggesting that the lack of CD209 expression (EBOVC) and differential regulation of LCK expression (KGC) is critical for host antiviral competence. However, since KGC possessed both genes, and showed statistical difference with EBOVC, understanding viral routes critical to LCK will be important in understanding EBOV pathogenesis in its host. The PCA analysis also showed the role played by LCK and IL2 in immune activation, given that the former was far from the correlation circle of unit radius and the latter was at the border of the circle, suggesting a high deviation from immune related function with possible interaction in a pathway that requires proper immune activation. LCK and other signaling genes, including TYRO3 [a receptor kinase involved in signal transduction from extracellular to intracellular matrix and also the PI3K pathway (Shimojima et al., 2006)], TBK1 [a serine/threonine kinase involved in regulating inflammatory responses from foreign agents, like virus and bacteria, which acts downstream of Toll-like receptors (Buss et al., 2004)] and IL1B (a potent inflammatory cytokine involved in prostaglandin induction and the activation of neutrophils, T- and B-cells) were out of the correlation circle, which suggests that they may all be involved in a similar immune pathway for virus clearance. This pattern of genes out of correlation was also observed in the silhouette plot, where some genes in the second cluster were completely on the negative side of the silhouette axis. Observations from this study suggest a viral selective process to better engage host cell receptors for better penetration and infection by subduing the MHC class I HLA molecules necessary for antiviral functions. Deciphering this pathway is very critical for understanding and differentiating viral related clinical manifestations and identifying the right genetic approach to arrest viral epidemics in the future.
This is the first study carried out to understand viral differential usage of host cell routes to penetrate and infect the cell. Relative to EBOV, for which a lot of scientific research is currently going on to identify a possible therapy to prevent future occurrence and also arrest the disease in case of an epidemic, other viral diseases show similar manifestations and use similar entry routes and cellular pathways to infect the cell and could be good controls in understanding genetic mechanisms of viral pathogenesis. The three main clusters showed clearly that viruses differentially regulate LCK in their related cells of interest. KGC shows that viruses directly infect immune cells mutating LCK and for MDC, viruses downregulate LCK hence escaping MHCI pathway and directly infecting B-cells while in EBOVC, LCK is expressed in dendritic cells and mutated with inhibition of MHCI pathway. Several transcripts which could be critical to LCK differential functional pattern in either the innate immune pathway or the Adaptive immune pathway were identified and understanding their role in proper immune activation is critical because viruses all escape the MHC class I peptide recognition pathway hence infecting immune T and B-cells. The differential expression of LCK in dendritic cells and immune cells suggest pathways required for complete immune activation and clearance of viruses, which is under viral control in infected cells. This mechanism results in poor transcription of LCK through differential promoter usage and prevention of proper LCK pathway activation for immune induction. Viruses included in this study could actually be classified into three groups, as follows: CD209 dependent (MDC), LCK dependent (EBOVC) and CD209 and LCK dependent (KGC). The differential regulation of LCK for proper T-cell induction through differential promoter usage in EBOV disease, are currently under research by our group. Markers to identify and classify viral disease types in humans based on CD209-LCK dependent pathways are currently being exploited.
RMEAN extractor software available from:
https://github.com/brack123/GEO_RMEAN_Data/tree/V.1.0.0
Archived source code as at time of publication: doi, 10.5281/zenodo.166757 (Achinko, 2016)
License: GNU
Dataset 1: Ebola related virus dataset in different hosts: Total sample data collected for this study, based on keywords “glycoprotein” and “virus”, doi: 10.5256/f1000research.9778.d143382 (Achinko et al., 2016a). The data are available in a .txt or .xls file. RSTD, relative standard deviation; RMEAN, relative mean; GBACC, gene bank accession; gdsType, dataset type.
Dataset 2: Ebola related virus dataset in human host: Titles and ranking of all 54 samples obtained for this study prior to filtering for humans, doi: 10.5256/f1000research.9778.d143384 (Achinko et al., 2016b). GP-V_Sample_1, glycoprotein and virus sample 1; GEO, Gene Omnibus.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)