Keywords
Ctenophora, Phylogenetic reconstruction, Ribosomal subunits, Non-fluorescent protein (GFP-like), Bayesian Inference, Maximum Likelihood, Isopenicillin-n-synthase (IPNS)
This article is included in the Phylogenetics collection.
Ctenophora, Phylogenetic reconstruction, Ribosomal subunits, Non-fluorescent protein (GFP-like), Bayesian Inference, Maximum Likelihood, Isopenicillin-n-synthase (IPNS)
The relationships among deep metazoans (Cnidaria and Ctenophora) and Parazoa (Porifera and Placozoa) are not totally clear1. In this paper we try to reconstruct the phylogeny inside the phylum Ctenophora with state of the art methods and compare our results with previous work2,3. Ctenophores are a key phylum for the understanding of the development of organ systems, triploblastic animals and bilateral symmetry4. Our goal was to reconstruct the phylogeny of previous studies using as many sequences as possible available on GenBank. The use of these sequences allowed us to perform a multilocus analysis (MLSA), instead of the single gene analyses previously performed. The sequences selected for this study have never been used for phylogenetic analysis exclusively of this phylum5,6.
Our research consists of 1) the analysis of ribosomal genes (5.8S; 28S; ITS1; ITS2 and 18S) and 2) the analysis of two ortholog genes found in ctenophores (a GFP-like non-fluorescent protein, and isopenicillin-N-synthase FYY1).
The ribosomal genes were analysed using partitioned nucleotide substitution models while the ortholog genes were analysed using partitioned amino acid substitution models. We compared the findings of our two approaches (ribosomal and ortholog genes) to each other and against the previously reported phylogenetic trees obtained from molecular data3 and morphological data2.
All sequences corresponding to Ctenophora (Taxonomy ID: 10197) were retrieved from GenBank’s nucleotide database. Short sequences (those shorter than 150 base pairs) or ambiguously labeled sequences (those not assigned to a specific species) were discarded. This criterion was used to obtain an almost complete matrix including most loci available of reported taxa across the phylum.
Seven loci were chosen for analysis; five corresponded to ribosomal RNA regions (5.8S; 28S; ITS1; ITS2 and 18S). The other two corresponded to ortholog genes: a putative non-fluorescent protein (GFP-like protein), and isopenicillin-N-synthase FYY1 (IPNS), a protein involved in the bioluminescence process. The sequences were extracted using the annotation of the retrieved records using Biopython 1.677.
The taxa present for each analysis is listed in Table 1 and Table 2. All sequences used (with corresponding accession numbers) and scripts used for analysis are available at http://doi.org/10.5281/zenodo.19308016.
(1) Sequence was available on GenBank. (-) sequence was not available on GenBank or not reported.
(1) Sequence was available on GenBank. (-) sequence was not available on GenBank or not reported.
Given the phylogenetic distance between the different taxa of this phylum, for the protein coding genes, we decided to work at the amino acid sequence level due to high sequence saturation at the nucleotide level.
The sequences corresponding to the ortholog genes were translated in silico using DNA2PEP 1.18 with standard genetic code, and aligned using MAFFT 7.2229. A MLSA was performed using these two loci. Alignments were concatenated using Python scripts and partitioned by gene to be analyzed for amino acid model and best partition scheme using PartitionFinderProtein 1.1.110 Model adjustment was assessed using Bayesian information criterion (BIC). The best model found by PartitionFinderProtein 1.1.1 for IPNS partition was LG + G + I, and LG + G had better adjustment for GFP-Like partition. Phylogenetic reconstruction for the ortholog genes was carried out by maximum likelihood (ML) and Bayesian inference (BI) methods, using both Garli 2.0111 and MrBayes 3.2.612, with the proper amino acid substitution model parameters for each partition.
For the ML analysis, using Garli, a total of 5 independent ML searches were performed and supported with 65 bootstrap pseudoreplicates. For BI analysis, using MrBayes, two independent MCMC runs (four chains for each) were carried out for 1.000.000 generations, using a relative burn-in discard of 35% of total sampled trees (sampling frequency of 100 generations).
For the five rRNA loci, the automated pipeline PhyPipe13 (available at: https://gitlab.com/cibiop/phypipe/) was used for phylogenetic reconstruction by BI and ML methods. A regular PhyPipe run comprises DNA sequence alignment with MAFFT 7.222, partition analysis with PartitionFinder, and phylogenetic reconstruction with RAxML 8.2.814, MrBayes 3.2.612 or Garli 2.0111. For this analysis, MrBayes was executed under the following parameters: two independent MCMC runs, four chains, 1.000.000 generations, 35% of relative burn-in and sampling frequency of 100. For ML analysis, Garli was executed doing first a ML search (5 independent searches), then 1000 bootstrap pseudoreplicates were performed and mapped to the best ML topology using SumTrees from DendroPy 4.1.0 package15.
The majority of the phylum analysed in this study show a standard grouping condition; the organisms that are related in one of the analyses are also related in the other. This is more evident comparing at family level, where the individuals of the same family, and in some cases order, grouped with other organisms of the same order. Exceptions are discussed below.
In the reported trees there are families represented by several species while complete orders are represented by just one species. For the purpose of clarity, from now on the families represented by several species will be discussed at the family level while the orders represented by just one species will be discussed using the representing species.
Thalassocalyce inconstans and Lampocteis cruentiventer are unexpectedly grouped together in both analyses, but the position is not the same in comparison with the other clades. The support values in the ribosomal tree are very low compared to the ortholog genes tree.
The order Cydippida is divided in five different clades or subgroups, shown in different tones of blue in Figure 1. We confirm that this group is paraphyletic as reported previously in 2,3. The species Bathyctena chuni (Bathyctenidae, Cydippida) is grouped with Ocyropsis maculata (Ocyropsidae, Lobata) in amino acid analysis, but there is no information on the ribosomal sequences of Bathyctena chuni, so it was not possible to compare.
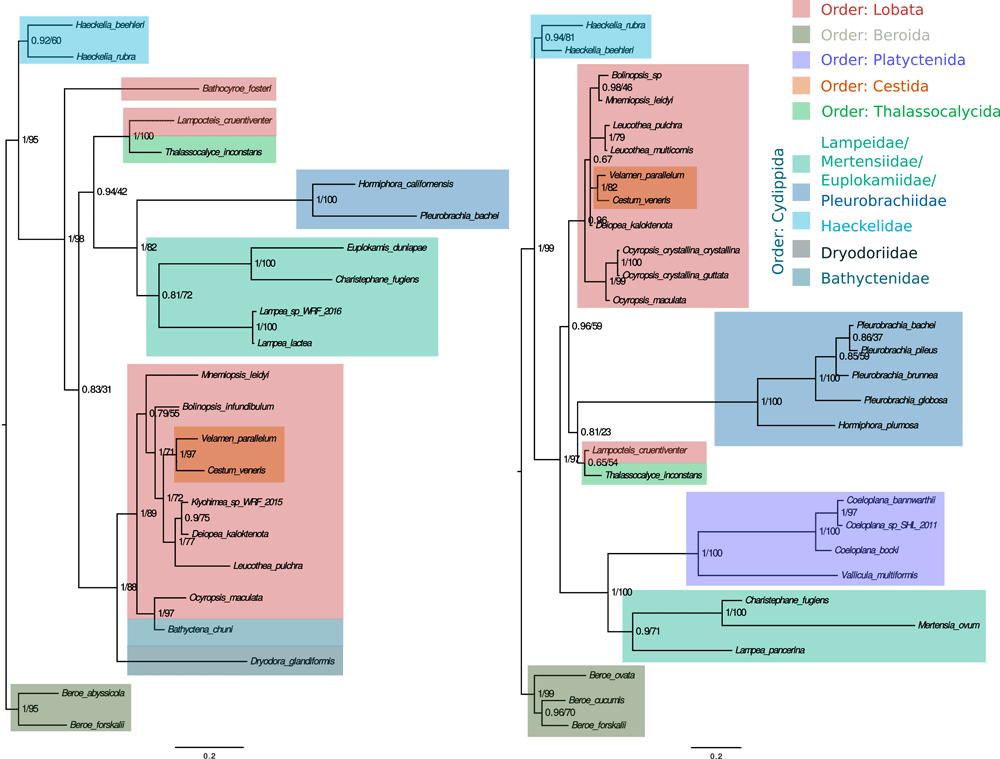
Trees were constructed using Bayesian inference and maximum likelihood methods and consistent topologies were found within methods. Support values are shown at nodes in the form of posterior probability/bootstrap value.
In the amino acid tree, Pleurobrachiidae, Mertensiidae, Lampeidae, Euplokamididae form a clade; but the relationships between them are not clear, and the bootstrap values and posterior probability are low in this group. In the ribosomal analysis we could include the Platyctenida order, which grouped with high support in the clade formed by the mentioned families and order. In the ribosomal analysis, we see some shared features with one of Harbison’s trees2. Also the Thalassocalyce-Lampocteis clade is related to this group, but the position varies depending of the analysis. Dryodora glandiformis groups with the clade formed by Lobata species, but this result is only evident in the ortholog genes tree, due to the lack of rRNA sequences for this particular taxon.
All the trees were rooted using Beroida as the outgroup, following the hypothesis that this is the most basal group. The same choice of root was made by Harbison2. Additionally, the Beroe genus is a good outgroup because it belongs to the class Nuda while the other studied species belong to the class Tentaculata (our ingroup).
Bathocyroe fosteri is present in an unexpected position in the tree. It should have been included in the Lobata clade. Instead, it was placed outside the subgroup containing the Lobata, Pleurobrachiidae, Mertensiidae, Lampeidae and Thalassocalycidae families. This finding is not compared with rDNA loci analysis since ribosomal sequences for Bathocyroe fosteri were not available.
In the research performed by Harbison2, the Lobata group was placed below the Cestida group. Later, this finding was not discussed by Podar et al. 20013 as in the ribosomal data they used both groups are in a polytomy. Our finding, using rDNA, is in concordance with the findings of Podar et al. 20013, but using the ortholog genes the finding is contrary to what was proposed by Harbison. We found, on the ortholog genes analysis, that Cestida group is the one derived from Lobata and not vice versa as suggested by Harbison. This finding has a high bootstrap value and posterior probability support.
The Haeckelidae family preserves its position in the phylogenetic trees placed as sister group of all the other Tentaculata taxa analysed, with high support, according to previous studies2.
The lack of reported DNA sequences of few groups, like the orders Crytolobiferida, Cambodjiida, Ganeshida; several families, like Eurhamplaeidae and some records reported as Ctenophora incertae sedis (Tentaculata incertae sedis), make it harder to have an entire vision of the phylogenetic relationships inside the phylum. The order Ganeshida is grouped with Lobata, according to Harbison2 and the lack of this group may have caused a misplacement of the Thalassocalycida representant.
To improve the results, Coeloplanidae should be included in the protein phylogenetic analysis. Further studies including more sequences from families such as Leucotheidae, Lampoctenidae and Thalassocalycidae are also needed to solve the resulting polytomies and to obtain better support to confirm the relationship between Thalassocalyce inconstans and Lampoectis cruentiventer in the rRNA analysis.
The raw data and scripts used for this project are available in Zenodo, DOI 10.5281/zenodo.19308016.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
While I am always glad to see new studies on ctenophore phylogeny, I am very surprised that you did not cite Simion et al. 2014 (of which ... Continue reading Dear authors,
While I am always glad to see new studies on ctenophore phylogeny, I am very surprised that you did not cite Simion et al. 2014 (of which I am the first author) for two reasons :
- You used all the data sequenced in that study.
- Both study are very similar in topic and design, and should therefore be compared.
Please find a link to the study : http://www.sciencedirect.com/science/article/pii/S0944200614000816Sincerely,
Paul Simion
While I am always glad to see new studies on ctenophore phylogeny, I am very surprised that you did not cite Simion et al. 2014 (of which I am the first author) for two reasons :
- You used all the data sequenced in that study.
- Both study are very similar in topic and design, and should therefore be compared.
Please find a link to the study : http://www.sciencedirect.com/science/article/pii/S0944200614000816Sincerely,
Paul Simion