Keywords
NCBI, dbVar, Structural Variation Cluster, GVF, Genomics, Open-Source, Genome Annotation, Education, Software
This article is included in the Bioinformatics gateway.
This article is included in the Hackathons collection.
NCBI, dbVar, Structural Variation Cluster, GVF, Genomics, Open-Source, Genome Annotation, Education, Software
There is a growing body of evidence suggesting that genomic structural variants play an important role in the etiology of human disease and in determining individuals’ characteristics and phenotypes1,2. Structural variants are also important for understanding the evolution of species3. dbVar is a database of large structural genomic variants that catalogs millions of records from both small and large studies and makes them freely available to the public4. The data are organized by submitted study, which makes for convenient comparisons between cases and controls. dbVar online search and browser tools make it easy to search and retrieve the data.
It is difficult to annotate novel SVs or to compute summary data without a reference record or exemplar when multiple SSV choices are available in the same genomic region, and there has been no publicly available resource to date that combines variants from all studies for integration into a bioinformatic pipeline for search, analysis, and comparison. We created structural variant clusters (SVC) to overcome these problems. Structural variant clusters (Figure 1) are smaller discrete genomic features that include counts of the features shared between SSVs. In regions with fuzziness between overlapping SSVs, SCVs allow the calculation of annotation and frequency by either consensus overlapping regions or by user-defined limits.
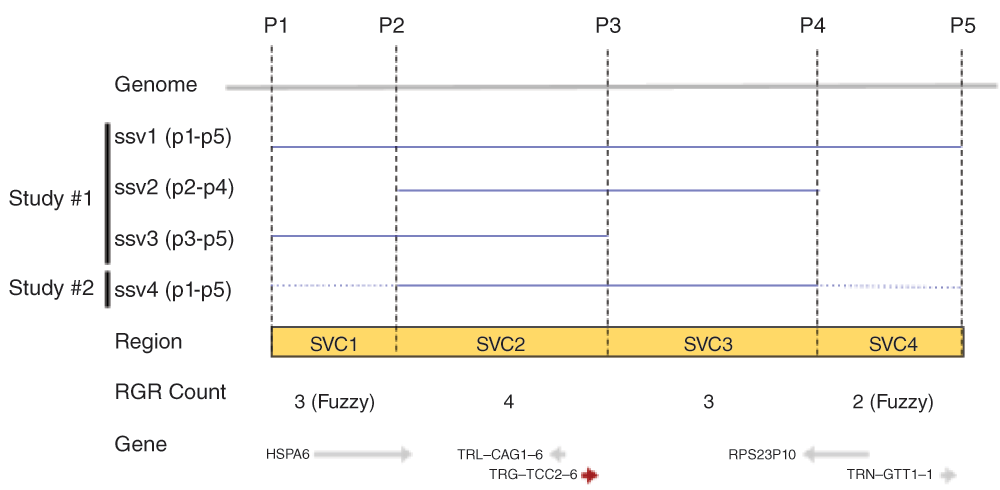
Reference genomic regions SVC1-SVC4 (yellow box) are demarcated by overlap and non-overlapping positions (P1-P2, P2-P3, etc.) between SSVs. The observed SVC counts and the genes are shown on the bottom.
Additional benefits of having a defined set of SVCs include:
improved data exchange, data mining, computation, and reporting;
better searching and matching of genomic coordinates across studies;
easier aggregation of annotations such as disease and phenotype, frequency, and genomic features that co-locate with a SVC;
a simplified display in the Sequence Viewer as an aggregated histogram or density track from all studies (currently dbVar display each study as a track, which can be slow to render and difficult to display on small screens); and
the ability to measure SSV concordance regions and validate across studies.
The Structural Variation Cluster project aimed to accomplish a number of goals. First, we generated a Genome Variant Format (GVF) file of SVC regions as defined above, based on RefSeq GRCh381. Each region is assigned a unique ID (SVC1, SVC2, etc.). The SVC VCF file is used as the basis for generating aggregated data, filtering, generating sequence viewer tracks, and for comparison with user data. We also generated a histogram track to show the frequency of the regions across studies in genomic context for the Sequence Viewer. In addition, we annotated SVC regions with Gene, colocated dbSNP reference SNPs, ClinVar, and other colocated features. We aimed to create a tool for filtering SVC GVFs by variant type, region size, region count, chromosome, and additional user-defined splitting and filtering parameters. This tool would allow users to compare their data with SVC GVFs and report matching regions of overlap.
SVCs are defined as the union set of overlapping and non-overlapping regions for all SSVs aligned to the genome using HTSeq version 0.6.05, based on the genomic coordinates in RefSeq human genome assembly GRCh38 (RefSeq accession GCF_000001405.26)1 (Figure 1).
Figure 2 demonstrates the workflow for this analysis. dbVar SSV data by studies were obtained in tab delimited format from the FTPsite (ftp://ftp.ncbi.nlm.nih.gov/pub/dbVar/data/Homo_sapiens/by_study/) and used as input. The study files were combined and sorted by chromosome positions into a single file using the script merge_data.py. SVC regions, including counts as shown in Figure 1, were generated from the merged file using the script make_gvf_and_bedgraph.py, which output SVC GVF and BED files. Since the approach in Figure 1 is similar to finding consensus regions or overlapping features between aligned reads make_gvf_and_bedgraph.py use HTSeq.GenomicInterval class to store SSV chr. start, and stop coordinates as genomic features and the HTSeq.GenomicArrayOfSets class to identify overlapping positions to generate SVC and counts.
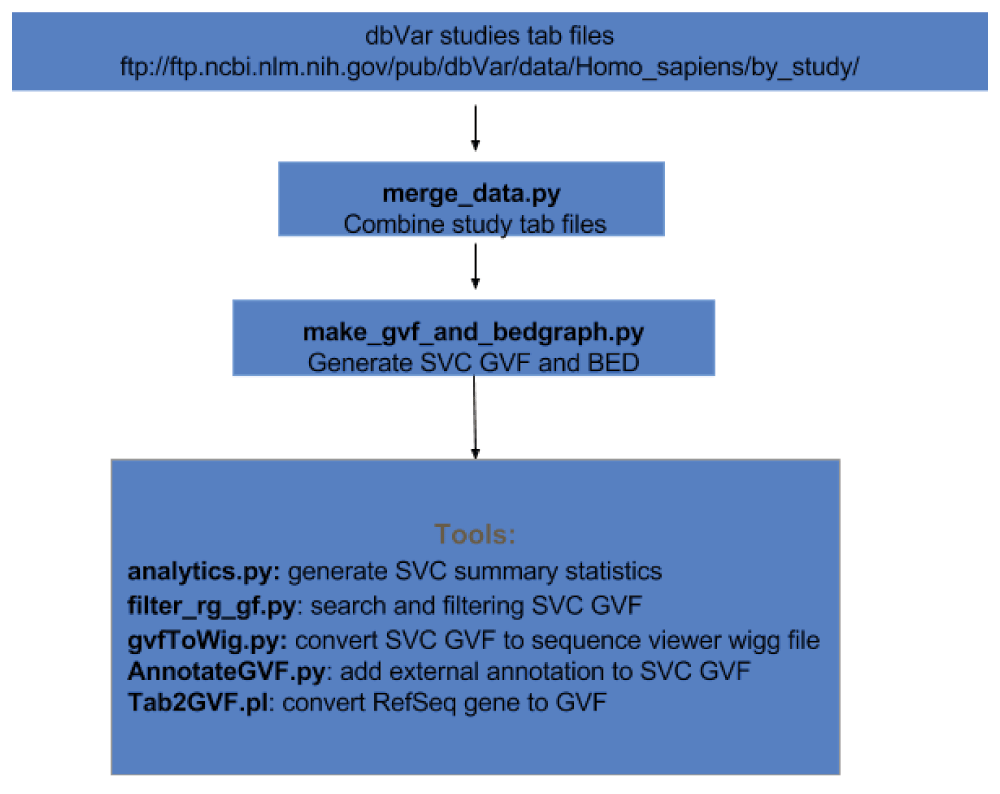
Additional tools are available as scripts using SVC GVF as input to compute summary statistics, to search and filter, to generate WIG files for viewing in sequence viewer, and to annotate using external data sources. All scripts and examples are available on GitHub (https://github.com/NCBI-Hackathons/Structural_Variant_Comparison/). For this study all coordinates reported are based on GRCh38.
As shown in Figure 1, SVCs were created from overlapping and non-overlapping regions of two or more SSVs using the HTSeq.GenomicArrayOfSets class and output as GVF file format. Each SVC is counted for the number of times it is present as a subregion of a SSV, providing a total SVC count across studies. A single SSV by itself without any overlap between itself and another SSV in the region constitutes a single SVC with a feature count of 1. 3.6 million dbVar SSVs generated 3.4 million SVCs for all dbVar data (combined-set) by variant type (Table 1).
The most common variant type was deletion followed by CNV. All CNV types combined (rows 1, 7, and 8 in Table 1) total 972,335. We also generated SVC for each variant type (ie. CNV, in/del, etc.) and by individual studies (study-set) for QA/QC and analysis between types and studies of interest. The study-set generated a total of 2.5 million SVCs versus 3.4 million SVCs from the combined-set.
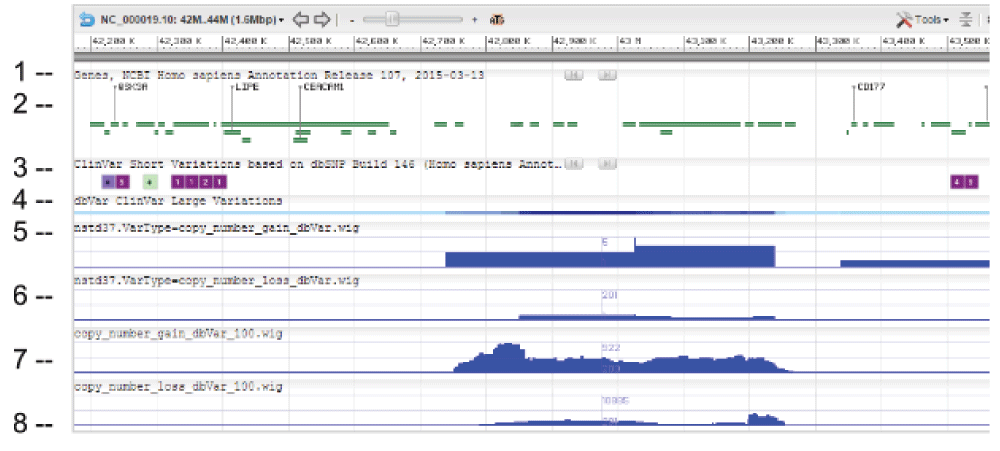
WIG files were generated from SVC GVF files to allow loading into sequence viewer for quick visual inspection as shown in Figure 3. The SVC sets used for inspections are the combined-set which includes 1000Genomes6, as well as other large studies to provide frequently occurring or “common” SVC to compare with presumed curated variants that have clinical significance from study-set (dbVar:nstd37) submitted by ClinGen7. The Variation Viewer8 allows for quick navigation by genes, chromosome positions, and variations for visual comparison (Figure 3, Figure 4, and Figure 5). Figure 3 and Figure 4 show a hotspot peak A in ClinVar (track 4) that corresponds with a peak in SVC from nstd37, suggesting that this region is critical for function and that variations in this region are rare. These conclusions are supported by the lack of corresponding SVC peaks in the combined-set “common” tracks 7 and 8. However, tracks 7 and 8 also contain peaks B and C that flank the ClinVar peak, which may demarcate the boundaries for the critical region peak A. In contrast, Figure 5 shows that there are corresponding SVC peaks in the nstd37 (rare) and in the combined-set (common), suggesting that variants in this region may have minimal or no clinical impact by themselves.
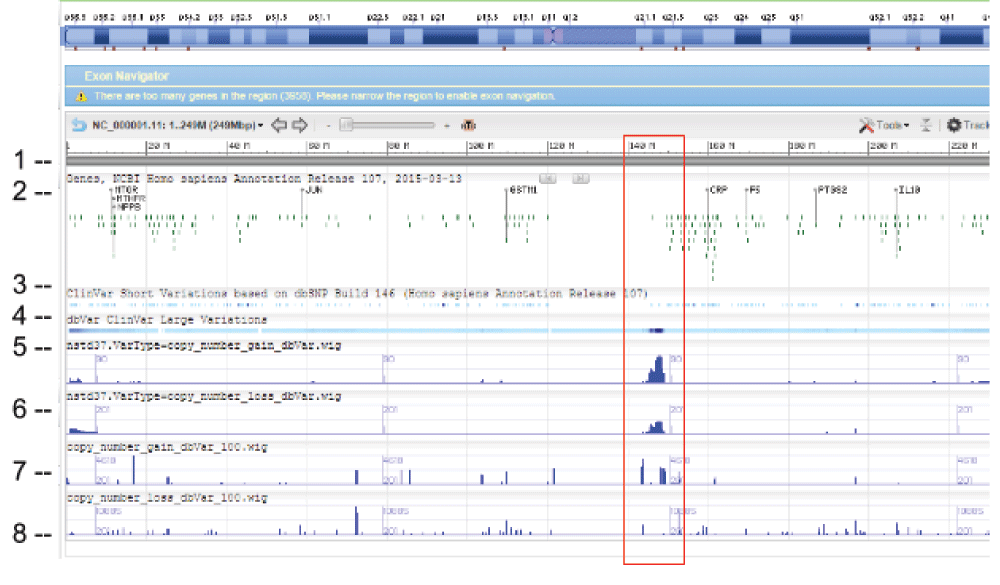
Starting from the top: (1) chr 1 sequence, (2) Gene track, (3) ClinVar short variation for dbSNP SNV, (4) ClinVar large variation, (5) ClinGen SVC study-set (dbVar:nstd37) copy number gain, (6) ClinGen SVC study-set (dbVar:nstd37) copy number loss, (7) SVC combined-set for copy number gain with count >= 100, and (8) SVC combined-set for copy number loss with count >= 100. The red box highlights an SVC hotspot region found in ClinGen (dbVar:nstd37) tracks 5 and 6 that correspond with the variants in ClinVar. The scale for SVC count histogram are 1–90 (track 5), 1–20 (track 6), 1–4618 (track 7), and 1–10885 (track 8).
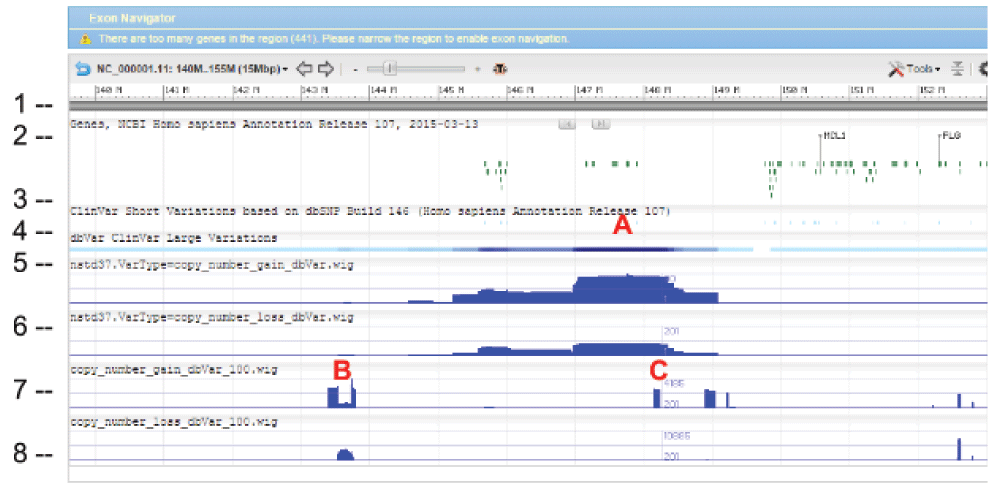
The tracks and histogram scales are as described in Figure 3.
The software tools we developed and provide here compute SVCs and provide counts of concordance regions across SSVs. We also developed tools to search, filter, annotate, and graphically view the results in sequence viewers or to incorporate them into custom analysis pipelines. Using these tools, we provide examples (Figure 3) for comparing across different SVC data sets with other annotation (such as genes and ClinVar). Such comparisons will allow users to investigate across the genome - or near a gene of interest - and to look for concordance and conflicts between data, which may help users form hypotheses regarding the biological impact of observed variation in SVC regions. In future, we will conduct the work and analysis required for SVC data quality assurance. We believe that SVC data promise to improve the analysis and the elucidation of the biological impact of structural variants, and in future, will probably have uses beyond those described here. Potential uses for SVC data could include:
the evaluation of other SVC hot spot regions to determine if they occur biologically or are due to genome problem regions;
the use of study metadata to validate SVCs that are in concordance with regions across studies and different assay platforms;
the validation of rare SVCs (count =< 2) and common SVCs ( count > 2);
identification of evidence of variations in all public SRA data;
combined analysis and annotation of SVCs to ClinVar, dbSNP, and other variation resources;
the creation of a reference dbVar “SV” number based on SVCs, which would be the equivalent to dbSNP’s RS number;
identification of population-specific SVCs to gain insight into the functional significance of structural variants and their evolution; and
determination of high-priority SVCs with significant functional impact and effects.
In addition, a “dbVar Beacon Service” could be developed to allow users to query dbVar if variants exists for a genomic location of interest using combined SVC data. The results would report the number of SVCs and associated SSV IDs and study IDs. Users could then download the study or SSV of interest from dbVar.
Latest source code: https://github.com/NCBI-Hackathons/Structural_Variant_Comparison/
Archived source code as at time of publication: http://dx.doi.org/10.5281/zenodo.482019
Accompanying wiki: https://github.com/NCBI-Hackathons/Structural_Variant_Comparison/wiki
Manual: https://docs.google.com/document/d/1WBnEnShnw28ZFg17A3xUpWOyvxXjb2q-h1kF-XYVWEw/edit?usp=sharing
License: CC0 1.0 Universal
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)