Keywords
Huntington's disease, gene expression, CpG-islands, caudate nucleus, chromatin regions
This article is included in the Bioinformatics gateway.
Huntington's disease, gene expression, CpG-islands, caudate nucleus, chromatin regions
Huntington’s Disease (HD) is a complex disease of the brain associated with massive changes in gene expression. The genetic cause was identified in 19931, but no successful treatment has been found yet. HD is a dominantly inherited neurodegenerative disease that affects 1 – 10/100.000 individuals, making it the most common heritable neurodegenerative disorder2.
Although HD is considered a monogenic disease3, extensive research since 1993 into the underlying pathology suggests that the disease mechanisms are more complex than originally considered. Transcriptional dysregulation is a widespread phenomenon in HD that can be observed well before the first clinical symptoms appear4. It suggests that mutant huntingtin causes a broad and complex cascade of downstream effects. There are several ways in which mutant huntingtin can interfere with the transcriptional machinery and alter gene expression5,6. Given the extent of the transcriptional changes, the question arises if epigenetic mechanisms that operate at a genome-wide scale are also involved in HD. There is increasing evidence for epigenetic mechanisms playing a role in HD in human and model systems. For instance, earlier computational analysis of HD gene expression data showed that expression is deregulated in large genomic regions, indicative of a coordinated genome-wide mechanism7. These studies did not determine whether genome-wide alterations in gene expression are associated with changes in the composition of histone modifications.
More recently, H3K4me3 was shown to be enriched in 136 loci in an HD case/control study, which included genes that may affect the neuronal epigenome at large8. In HD mice, down-regulated genes were found to be associated with a selective decrease of H3K27ac and RNA Polymerase II9. Inhibitors of HDAC1, broad-range regulators of chromatin structure, have indeed been shown to be effective in HD mice10. The interplay of enzymes involved in post-translational histone modifications like histone deacetylaces (HDACs), may make chromatin less accessible in many places and therefore alter gene expression patterns. In Drosophila, the homolog of htt was found to suppress position effect variegation (PEV), possibly by influencing PEV modifier genes11.
A role for epigenetic mechanisms in HD is further corroborated by numerous neurodevelopmental and neurodegenerative disorders that have been associated with an altered chromatin structure12–14. Neuroepigenetics has therefore become a prime topic of interest15, with researchers seeking to identify epigenomic signatures and how they can contribute to brain health or brain disease16.
Considering the growing body of evidence that epigenetic regulation is involved in neurological pathology, we asked if the massive transcriptional dysregulation in HD coincides with large scale changes in chromatin state across the genome. Under that assumption, we hypothesize that significant numbers of differentially expressed genes in HD will be found in regions that are not normally associated with active chromatin states.
Here we report on a computational test of our hypothesis before laboratory experiments using only publicly available data sets: HD gene expression from the Gene Expression Omnibus (GEO)17 and chromatin state from ENCODE18. We assessed the overlap between differentially expressed genes and chromatin state, and we applied literature-based concept profile analysis (CPA) to interpret our findings19,20.
Our results indicate that the massive transcriptional dysregulation in HD is not matched by a significant largescale change of activity of genes that are part of inactive chromatin states in reference tissue. Our report includes a functional characterization of differentially expressed genes in HD in relation to huntingtin, chromatin state and CpG islands, based on a literature-based semantic analysis.
The analysis we performed to test our hypothesis is part of an interdisciplinary research approach where computational analysis is used to help steer laboratory experiments to increase the overall efficiency of a research laboratory21.
In Concept Profile Analysis (CPA), the vector space model is used to associate two concepts mined from the literature with each other. Advantages of this model include efficient and transparent comparisons, and the possibility of attaching a weight to the association20. The CPA algorithms have previously been used for a range of different gene expression data analysis purposes such as functional annotation22, comparison of studies23, prediction of novel interactions24, generation of gene sets19,25, and association with chemical structures26). The methodology has been described previously27. In short: In our database, every concept is associated with PubMed records using the indexing engine Peregrine (https://trac.nbic.nl/data-mining/) which is equipped with an in-house thesaurus of biomedical and chemical concepts that have been prepared for text mining28,29. For all concepts except genes and Gene Ontology (GO) terms the PubMed records are comprised of the texts in which the concept is mentioned. For genes, only a subset of PubMed records are used in order to limit the impact of ambiguous terms and distant homologs. GO terms are sometimes given as words or phrases that are infrequently found in the normal texts. To still provide broad coverage of GO terms, the PubMed records that were used as evidence for annotating genes with the GO term are added. For every concept in the thesaurus that is associated to at least five PubMed records, a vector containing all concepts related to the main concept (direct co-occurrence), weighted by the symmetric uncertainty coefficient is created. We call this a "Concept Profile". Concept profiles are matched to identify similarities via their shared concepts (indirect relations). Any distance measure can be used for this matching such as the mutual information, inner product, cosine angle, Euclidean distance or Pearson’s correlation. The CPA Web Services that we used for our analysis use an inner product measure30. These web services can be found in the BioCatalogue web service registry https://www.biocatalogue.org/services/3559.
For data analysis and interpretation we implemented a series of workflows using the Taverna workbench (Taverna workbench version 2.4.0)31,32. Taverna is an open source software for the development and execution of workflows. Our workflows are deposited online on the Zenodo repository (https://doi.org/10.5281/zenodo.16420133 and https://doi.org/10.5281/zenodo.16419834) and the myExperiment platform (http://www.myexperiment.org/packs/553).
The first Taverna workflow load_data_identify_DE_genes_Array_A, was implemented to examine differential gene expression between control and HD samples. Required workflow inputs were two data files with gene expression values and the phenotype information that describes the samples from the microarray experiment. Differential expression was computed using moderated t statistics with the package limma35 (version 3.14.1), which is provided by the bioconductor project36, R version https://www.bioconductor.org/. We analyzed each brain region separately, because previous analysis37 revealed regional patterns in gene expression. The workflow maps the expression data from probes to entrez gene ids using the Affymetrix Human Genome U133 set annotation data, (packages hgu133a and hgu133b, version 2.8.0; https://bioconductor.org/packages/hgu133a.db; https://bioconductor.org/packages/hgu133b.db/). When multiple probe names map to the same gene id, the ones exhibiting the most significant changes were used for further analysis. Final outcome of this workflow is a report. Each row is composed of a gene id, a fold change and its corresponding P-value indicating the significance of every change in gene expression, between HD and controls for each brain region. Adjusted P-values, generated by Benjamini and Hochberg’s method for multiple testing correction, are also included38. This workflow can be adjusted to compute differential gene expression between other variables such as male/female or the grade of disease pathology by editing the nested workflow “compute_DE_limma” within the R workflow component. An additional workflow create_exprs_obj_download_files is included in the myExperiment pack that was used to download data from the ArrayExpress repository. The workflow saves the gene expression data and the corresponding phenotype file in the directory indicated in the workflow input.
We note that this particular microarray experiment was composed of two microarrays, Human Genome U133A and U133B. For convenience we added the workflow: Get differentially expressed genes for Array B one brain region, but in principle the first workflow could be reused by adjusting the “libraries” component.
The second workflow map_genes_on_chromosome uses the output from the first workflow in order to map genes to their corresponding genomic location. The workflow uses the Biomart39 service within R, to obtain information regarding the position of each gene at the chromosome, HGNC gene symbols40, transcription start and end site and the transcription strand. The database that was used was the Ensembl genes 68 from Sanger institute and the Homo Sapiens dataset GRCh37.p8. The mouse assembly that was used to map genes to their chromosomal location was Dec 2011, GRCm38mm10.
The last workflow get_promoter_region_calculate_overlaps, first computes a promoter region for each gene and then operates on genomic intervals to compute gene promoters that overlap with a genomic region. The promoter region is computed for each gene, according to prespecified values, indicating the number of base pairs (bp) upstream and downstream of the transcriptional start site (TSS); for the CpG island analysis 5000bp upstream and 2000bp downstream and for the chromatin states analysis 50bp upstream and 50bp downstream was used. The decision for the promoter size in each case was taken after discussing with the domain experts and from knowledge acquired from previous experiments using the ENCODE data from Ernst et al.41. Using those data we performed multiple runs with different input values for the “upstream” and “downstream” variables, and the overlap parameter (Supplementary File 1).
When genes had multiple transcription start sites, we computed a promoter region for every TSS. Next part of this workflow is to compute overlapping regions between the input datasets. It includes a two sample Kolmogorov – Smirnov test to compare the empirical cumulative distribution functions (ecdf) of the P-values between the gene promoters that overlap with a specific genomic region and the ones that do not. The null hypothesis tested here was that there is no difference between the two groups. The workflow returns two lists of genes, one for the genes that overlap with a particular genomic region and another that does not. Furthermore, the results of the statistical test are reported: the ks test statistic (maximum distance D between the ecdf of the two samples) and the P-value of the test. If P-value < 0.05 we reject the null hypothesis.
The workflows that we implemented for gene interpretation and gene prioritization are based on the workflow pack at Zenodo, https://doi.org/10.5281/zenodo.164198 (see also, at myExperiment: http://www.myexperiment.org/packs/368), and are implemented using CPA web services30.
The CPA workflow Annotate gene list with top ranking concepts annotates a gene list with top ranking concepts by matching concept profiles of genes with for example in our case the concept set of Biological Processes. The web services part of this workflow query the Anni database27 that stores the concept profiles for each concept of interest. The first web service mapDatabaseIDListToConceptIDs maps a list of concepts, in our case Entrez gene identifiers, to their corresponding concept profile ids. Necessary inputs are a concept list (gene list) in a comma separated file, and the database identifier of the gene list necessary for the mapping (EG for Entrez Gene, see here: https://www.biocatalogue.org/soap_operations/41197 for more details on database identifiers). The next web service getSimilarConceptProfilesPredefined matches our gene list with the predefined concept set “Biological Process” (ID = “5”), and gives the top scoring biological processes that describe our gene list. For a complete list of predefined concept sets, the workflow List Concept Sets (provided in the current pack) can be run to choose the ID of the predefined concept set of interest. The web service getConceptName, gives the complete (human readable) names of the top matching biological processes. Lastly, the workflow Explain score between two concepts can be included to the analysis to provide evidence for the association between each gene and the annotations of the biological processes. The evidence reported is a list of concepts that link one concept with another and the contribution to the overall strength of the association. In addition, the corresponding concept ids are reported.
The workflow Prioritize gene list can prioritize a set of genes with respect to their association with particular concepts, in our case the HTT concept and epigenetics (concept profile: “epigene”). In order to obtain the concept profile identifiers the workflow getConceptSuggestionsFromTerm needs to run first.
Human brain data. The HD human brain data that was used in this analysis was originally produced and analyzed by A. Hodges and co-workers37. This experiment contains 44 HD positive cases and 36 age and sex matched controls. The processed data are available from the public repository NCBI Gene Expression Omnibus, entry GSE3790. Three brain regions were included and analyzed; the caudate nucleus, frontal cortex and cerebellum, with an Affymetrix Microarray GeneChip (Human Genome U133A and U133B). Furthermore, the HD positive cases were further classified based on whether symptoms were present or absent and according to Vonsattel grade of disease pathology (scale = 0 – 4). In our analysis we used the processed data and performed our own differential gene expression analysis (Dataset 142) with the workflow that performs differential gene expression analysis that was described previously in Methods.
CpG island data. CpG island information in the human genome was obtained from UCSC genome browser43, hg19 assembly FEB 2009 1 (Dataset 244). Here, CpG islands are marked as the DNA regions where the following conditions hold:
GC content of 50% or greater
length greater than 200 bp
ratio greater than 0.6 of observed number of CG dinucleotides to the expected number on the basis of the number of Gs and Cs in the segment
The ratio observed/expected (Obs/Exp) CpG was calculated as follows:
where N is the total amount of nucleotides in the sequence that is being analyzed. For CpG island information of the mouse genome the assembly dec 2011 (GRCm38/mm10) was used.
Chromatin states data. The chromatin marks were obtained from the encode project45. The chromatin states were part of an integrative analysis of 111 reference human epigenomes profiled for histone modification patterns based on DNA accessibility, DNA methylation and RNA expression. We used the two cell types that were more suitable for our analysis; the anterior caudate and dorsolateral prefrontal cortex (Dataset 346 and Dataset 447, respectively). The chromatin states we used for the current analysis were: active transcription start site proximal promoter (TssA), bivalent regulatory region (TssBiv), heterochromatin (Het) and repressed Polycomb (ReprPC).
Mouse data. The mouse brain data was taken from a published study10. There, total RNA was extracted from cortex, striatum, and cerebellum from WT and R62 transgenic mice. This study examines the effect of the HDACi 4b inhibitor on the disease phenotype. However only the data from animals treated with vehicle was used (Dataset 142). This study used Illumina Mouse Mouseref-8 Expression Beadchips v1. The raw data were analyzed using the Bioconductor packages and contrast analysis of differential expression was performed by using the LIMMA package. The differential expression values are available in the supplemental material of that publication.
To test if and how differential gene expression in HD is associated with particular chromatin states we used publicly available datasets from the GEO and ENCODE public repositories. We selected HD gene expression data from three regions of the brain made available by Hodges and coworkers37, and data from the ENCODE consortium carrying information about four chromatin states. These were: active TSS proximal promoter (TssA), bivalent regulatory region (TssBiv), heterochromatin (Het) and repressed Polycomb (ReprPC)45. Briefly, the first state pertains to active genes, the second to repressed genes that are ready to be activated, and the latter two represent repressed genes. The chromatin states were part of an integrative analysis of 111 reference human epigenomes profiled for histone modification patterns, DNA accessibility, DNA methylation and RNA expression. The different chromatin states were defined by a computational model that is based on a multivariate Hidden Markov Model48.
First we confirmed the results reported by the previous study37: large and numerous changes in transcriptional activity in the caudate nucleus brain region and notably smaller changes in the frontal cortex and cerebellum. More specifically, the number of differentially expressed genes was 5219, 127 and 96 for each brain region respectively with a FDR of 0.05. These results confirm previous observations illustrating that specific HD-affected brain regions exhibit defined changes in gene expression, in line with observable physiological effects37.
Secondly, we paired the data describing gene expression changes in the caudate nucleus and frontal cortex from the Hodges study to chromatin state data from the anterior caudate and dorsolateral prefrontal cortex from ENCODE. We considered these brain regions as the most comparable between the two studies. The cerebellum was excluded from this part of our analysis due to the absence of chromatin state data at ENCODE for the cerebellum. We determined the reference chromatin state of the genes from the gene expression study by determining the overlap of their promoter regions with the start and end positions of the chromatin state from the aforementioned brain regions used by ENCODE. The effect of a chromatin state on differential gene expression in HD was assessed by comparing the distribution of expression levels of genes overlapping with a particular chromatin state with the distribution of expression of non-overlapping genes. If chromatin state has substantial functional impact on the genes that are differentially expressed in HD, then these are expected to be significantly different. More details on the promoter region calculation and the overlap between the genomic regions can be found in the Methods section.
Specifically, we compared the distribution of the P-values for differential expression between these two groups of genes and assessed the difference by a KolmogorovSmirnov test, in the two brain regions and for each chromatin state (Figure 1). In the caudate region, we found an enrichment of genes overlapping with the active TSS and a depletion of genes overlapping with ReprPC (Figure 1). Heterochromatic or bivalent chromatin were not significantly associated with a genomic location of differentially expressed genes. A similar, but less pronounced pattern was observed for the frontal cortex for both the active TSS and ReprPC. In addition, in the frontal cortex the number of genes overlapping with the bivalent state was reduced. The magnitude of the association of a brain region with each chromatin state is in line with the HD neurodegeneration pattern, where the caudate nucleus exhibits the largest gene expression changes while the frontal cortex exhibits an intermediate to low pathology. In summary, we found enrichment of differentially expressed genes in the active chromatin state of reference tissue, but no strong evidence for significant enrichment in chromatin regions that are associated with repressed gene expression activity.
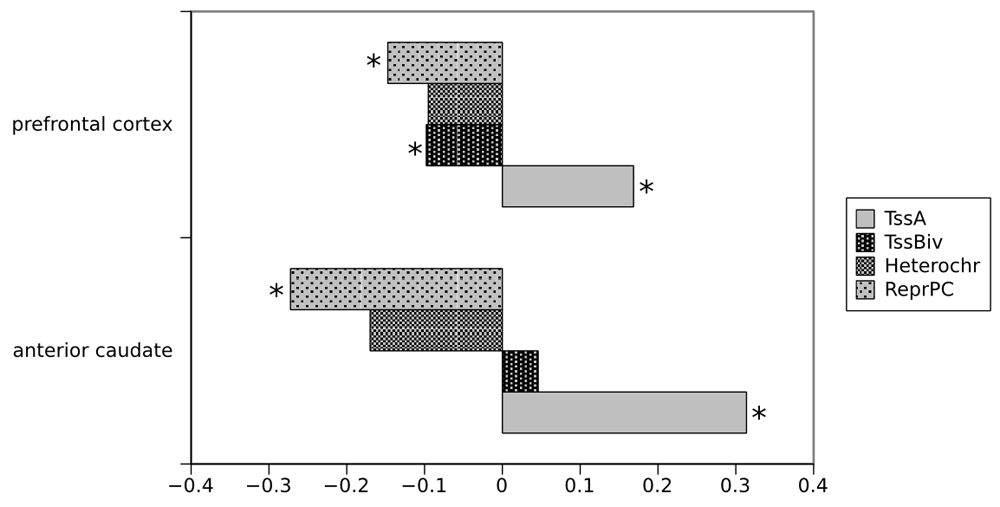
For each brain region, the P-value distribution for differential expression in HD patients compared to controls was compared between genes overlapping with a specific chromatin state and all other genes, in the anterior caudate and dorsolateral prefrontal cortex cells. The plot displays the maximum absolute distance D between the cumulative distribution function of the P-values for each group of genes (overlapping and non overlapping), as reflected by the KS test statistic. Positive values correspond to an enrichment of differentially expressed genes in a chromatin state, negative values with a depletion. Stars indicate a significant enrichment/depletion cf the KS test (P-value < 0.05). TssA= active TSS, TssBiv= bivalent Tss, Heterochr= Heterochromatin, ReprPC= repressed Polycomb state. For caudate: TssA D= 0.31, TssBiv D=0.04, Heterochr D=0.17, ReprPC D= 0.27, for frontal cortex TssA D=0.168, TssBiv D=0.097, Heterochr D=0.095, ReprPC D=0.147. The corresponding p-values were for the caudate : TssA pval < 2.2e-16, TssBiv pval= 0.59, Heterochr pval= 0.10, ReprPC pval=4.5167e-12, for frontal cortex TssA pval < 2.2e-16, TssBiv pval= 0.0002, Heterochr pval= 0.872, ReprPC pval= 1.4596e-05.
To further interpret the results from the chromatin state analysis, we used literature information (CPA19,27) to assess the biological processes within the lists of genes per chromatin state that were associated with gene deregulation (Dataset 549).
Using CPA, we found that for the caudate nucleus, genes in the active TSS state are mainly associated with processes related to transcription, cell cycle, protein transport and modification, RNA splicing, chromatin modifications and RNA processing. Genes in the repressed polycomb chromatin state are mainly associated with (brain) developmental processes and responses related to zinc and cadmium stimulus.
For the frontal cortex, genes in the active TSS state are associated with protein transport and modification, cell cycle, RNA splicing and signal transduction pathways (MAPK, notch, smoothened). Genes in the bivalent TSS are associated with brain development, neurogenesis, synapse and responses related to zinc and cadmium stimulus. We found that the genes that are part of the polycomb repressed group to be associated with similar functions as the genes in the bivalent state: (brain) developmental processes, responses related to zinc, cadmium and copper stimuli.
We note that at this point of our analysis, we filtered out genes of which the promoters were labelled with more than one state, using the criterion of at least 50 bps overlap in a promoter region of 100 bps. Interpretation of this set of genes would be ambivalent.
Because CpG island methylation is a known epigenetic regulatory mechanism that is ubiquitous in the human genome and may be a target for genome-wide regulation50,51, we also applied our approach to test if genes within CpG islands are overrepresented among differentially expressed genes in HD. We measured this by similar KS test statistics as in the previous section. We found that genes overlapping with CpG-islands in their promoter region, were significantly enriched in the group of HD-deregulated genes in all three brain regions (Figure 2A; P-value < 0.05), which supports taking this mechanism into account to formulate hypotheses when studying gene deregulation in HD.
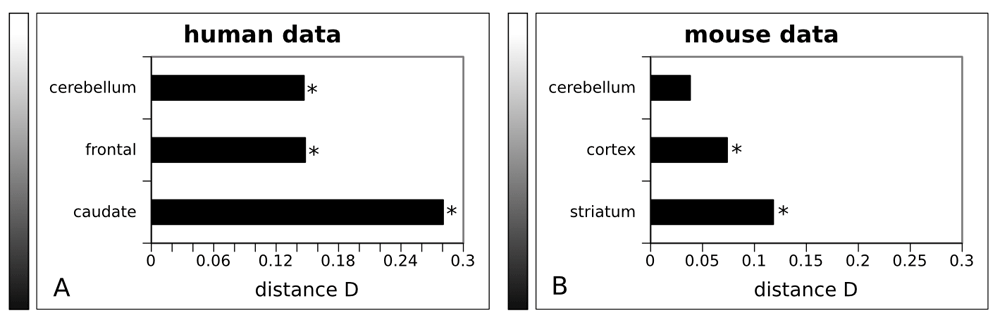
Maximum distance between the cumulative distribution function of the P-values between genes containing a CpG island in their promoter and genes that do not. The maximum distance (D) for each brain region was plotted for both datasets (human (A) and mouse (B) datasets). The gradient adjacent to each plot indicates the extent of neurodegeneration in each brain region. Black represents severe and white mild neurodegeneration. A: The analysis performed in the human data. Caudate nucleus is exhibiting the largest differences with a distance D between the two distributions of 0.2808, frontal cortex follows with D = 0.1482 and cerebellum with D = 0.147. B: The analysis performed in the mouse data. The results are analogous to the human data with striatum showing the largest differences with D= 0.118, cortex following with D=0.0739 and cerebellum with an insignificant difference (P-value = 0.596 >> 0.05) of D = 0.0383. * : depicts significance.
To further analyze and verify the association of gene deregulation in HD with the presence of CpG islands, we also analysed overrepresentation in data obtained from a HD transgenic mouse model10. Our results based on the mouse data corroborated those from the human data (Figure 2B). Comparable to the results for caudate nucleus in human data, the most highly affected mouse brain region, the striatum, showed the biggest overrepresentation of CpG islands among deregulated genes.
We next examined the biological processes that are associated with the differentially expressed genes that do or do not overlap with CpG islands by CPA. We inspected the top 50 annotations of each group. We identified many similarities between these two groups, but also annotations that were specific to each group (Dataset 652). For example, concepts related to chromatin alterations, such as chromatin remodelling and chromatin (dis)assembly, and histone post-translational modifications, such as (de)acetylation, and (de)methylation, were only found with a high rank in the list of genes containing CpG islands in their promoters.
Conversely, lymphocyte activation, angiogenesis, antigen presentation and neurogenesis were only high ranking associations for the non-CpG containing genes.
Some annotations were found in both groups but in a different ranking order. For example, the rankings of gene silencing, RNA splicing and phosphorylation were increased for genes within CpG islands while transcriptional activation and mitotic cell cycle rankings were higher for non-CpG containing genes (Dataset 652).
CPA can also be used to prioritize findings by a specific biological interest. In our case, we aimed to prioritise the list of genes overlapping with CpG islands that were also differentially expressed in HD caudate nucleus by their association with HTT and epigenetics. Here, we prioritised 100 proteins based on their association with HTT and epigenetics and absence of a direct relation with HTT by CPA (Dataset 753). Such relations are typically novel, or lost in tables or figures. If a novel relation is found (i.e. the relation is not found in our database of relations in MedLine abstracts), then CPA also provides intermediate concepts that link the two concepts.
In Table 1, we present the top 5 novel proteins that have the strongest association, and the intermediate concepts that link each prioritized protein to HTT and epigenetics. The intermediate concepts are grouped under the semantic categories “General”, “Biological Processes”, “Disease or Syndrome”, “Homo Sapiens proteins” and “Molecular Functions”.
Grouping the evidence provides a more complete insight into the processes involved in gene deregulation in HD mediated by CpG islands. For example, for one of our candidates, the amyloid beta (A4) precursor protein-binding, family A, member 1 (APBA1), we found nerve tissue (General), endocytosis (Biological Process), Alzheimer’s Disease (disease or syndrome), GRIN2B (H. sapiens Genes) and kinesin activity (Molecular Function) as intermediate links with HTT. This suggests that mechanisms involving APBA1 in HD share common components with the mechanisms involved in endocytosis and Alzheimer’s Disease and those involving GRIN2B and kinesin activity. Intermediate links with epigenetics were respectively: epigene (General), methylation (Biological Process), hypomyelination and congenital cataract (disease or syndrome), CDKN2A (H. sapiens Genes) and MGMT - O6-alkylguanine DNA alkyltransferase - (Molecular Function). Accordingly, these concepts provide suggestions about the epigenetic role of APBA1, which can be taken into account when further studying the role of APBA1 in HD.
We next investigated whether the prioritized genes reflect valid biological knowledge. Figure 3 shows that CPA is able to prioritize true associations with huntingtin as measured by a gene expression experiment, but that combining experimental (differential expression) measurements and literature evidence enables to select even more specific HD signatures. We used our concept profile technology to match all genes in our database that have a concept profile (12,391 genes) to the “huntingtin” concept profile (black line). We then compared the distribution of those CPA scores to the CPA scores of genes that were found to be differentially expressed in the caudate nucleus (p value < 0.05). We included two gene lists in our analysis: the top 100 most differentially expressed genes (red line) and the top 1000 (green line). The shift in the distribution of CPA match scores between the differentially expressed genes (top 100 and top 1000) and the scores of all genes reflects the added value of CPA (CPA scores of top 100 and top 1000 can be found in Dataset 854). We found a significant shift in the scores when comparing all CPA scores from our database with the top 100 differentially expressed genes: p = 2.67e −08 and the top 1000 p < 2.2e− 16.
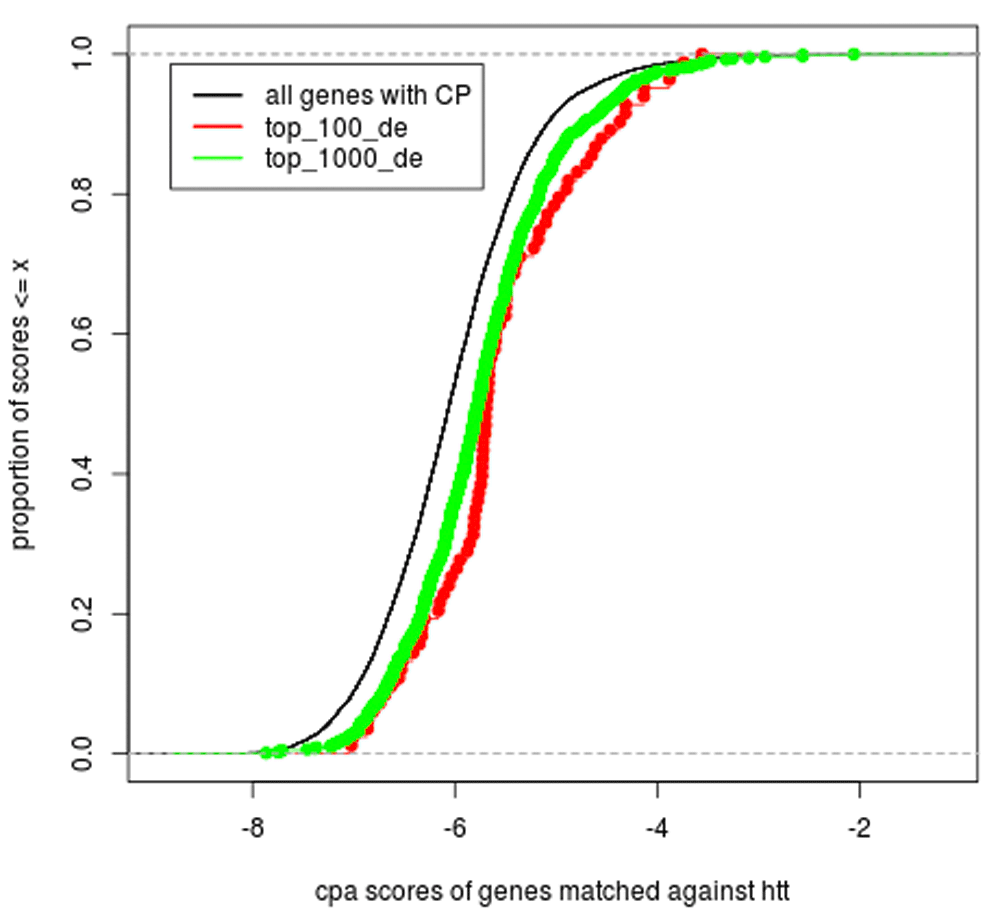
Cumulative distribution of match scores of the concept profiles (CP) between differentially expressed genes in HD with the concept profile of htt: match scores of all genes with a concept profile (black), match scores of the top 1000 differentially expressed genes (green), and the match scores of the top 100 of differentially expressed genes (red).
Also the top 100 and top 1000 differ significantly (p = 0.03184), showing that it is useful to narrow down on the top ranks for follow-up research. In principle, more extreme p-values are associated with higher CPA scores. In addition, to show that our list of 100 prioritized gene-HTT CPA match scores would not have been found by chance, we assessed the percentile score of our list when compared to the frequency distribution of 100 match scores of randomly sampled gene-HTT concept pairs out of the 12,391 genes in our concept profile database (Dataset 955). All genes were in the top 95 percentile, except NTRK3 (55 percentile).
The computational analysis with public data that we present in this paper shows that there is no strong evidence for genome-wide relocalization of gene activity to repressed chromatin states, at least not at a scale that could explain the massive transcriptional deregulation that we observe in HD. Most of the deregulated genes mapped to the active chromatin state of our reference tissue and were underrepresented in silenced states of chromatin (Figure 1). Previous reports supported the implication of large scale chromatin alterations in gene deregulation in HD. For instance, Anderson et al. reported that gene expression is deregulated in large genomic regions in blood and post mortem tissue of HD patients7. The authors inferred a relation with repressed and active chromatin, but did not use chromatin annotation data directly. Our results suggest that the association with genome clusters is mostly within the active state and does not extend to disruption of chromatin states at large scales.
Active chromatin is normally more prone to regulation and deregulation. Our results suggest that the epigenetic mechanisms that have been observed in HD are mainly bound to this fraction of chromatin. We speculate that the effects are more closely associated with transcription regulation of individual genes, than with large scale higher-order rearrangements of chromatin structure56. Our CPA analysis of deregulated genes with CpG-islands corroborates this suggestion: chromatin-related concepts from our CPA ranked high in this set of genes, while CpGislands are mostly known for their direct role in transcription initiation of the proximal gene. Nevertheless, CpG-mediated changes in gene expression are a common mechanism for many genes and deregulation of this mechanism is thus likely to have a genome-wide effect. It was unexpected that the differentially expressed genes are significantly depleted in the bivalent chromatin state in the prefrontal cortex. The bivalent state is expected to be associated with genes that are prone to become active. Our expectation was that deregulated genes in this brain region would be weakly enriched in this state, as we did find for the caudate nucleus. We currently lack a good explanation for this result.
We incorporated the computational analysis on public data in our research strategy to advise on next experiments. However, working with public data has its limitations. Here, we had to rely on reference tissue for the chromatin state in order to compare it with gene expression. In addition, the reference tissue chromatin state was measured in healthy individuals. Therefore, we cannot rule out that large scale chromatin effects will be observed when chromatin state and differential expression are measured together in new HD specific experiments. This could reveal new evidence for chromatin state deregulation in HD and give insight in the relationship with transcriptional deregulation, possibly at higher resolution than we could achieve in our current study. However, our results suggest that this is not very likely, leading us to advise fellow HD researchers to prioritise experiments that assess the role of epigenetic mechanisms in HD at the scale of individual genes or small clusters of genes, and within the active fraction of chromatin.
Our results can be used to refine hypotheses about molecular mechanisms involved in HD. For instance, we surmise that the reported association of DNA methylation and chromatin organisation, and the effects of HDAC on the HD phenotype in mice57 are bound to the active fraction of chromatin. Furthermore, it appears that CpG islands located within the promoter region of a gene increase the probability that genes in such genomic regions are deregulated in HD. This is in accordance with a study where changes in DNA methylation were observed in cells expressing mutant huntingtin58. This in turn suggests that DNA methylation in promoters is implicated in alterations in the brain, which is in accordance with a study that noted changes in DNA methylation in cells expressing mutant huntingtin58. Based on our semantic analysis chromatin remodelling, chromatin (dis)assembly, and histone modification were associated with altered gene expression profiles. In contrast, non-CpG containing genes are more likely involved in immune response and neurogenesis, which represents functionally linked processes59.
Furthermore, we used CPA as a means to prioritise genes for our hypotheses. For instance, we prioritised genes overlapping with CpG islands by their association with HTT, assuming that CPA rank scores are higher for genes that are higher-up in the cascade of events caused by the mutant protein. We recently showed that this is a fair assumption for CPA, although literature bias cannot be completely mitigated60. Similarly, we further prioritized candidate genes in terms of their association with epigenetics. Genes such as APBA1, KAT2A, CARM1, SLIT2 and BNIP3 came forward as the most likely candidates to play a role in HD in the context of downstream effects of HTT involving epigenetic mechanisms. These candidates were filtered on potential novelty: only those genes were reported for which there was no direct association found in our database of PubMed abstracts. This does not exclude associations that were reported in tables and supplemental material that are much harder to mine for technical and legal reasons. Our study also retrieved several well established associations consistent with earlier studies.
In summary, our results show how literature information in combination with data analysis present useful tools for exploration of hypotheses for possible future experiments.
Our methodology offers support for hypothesis generation to elucidate missing links in mechanisms involved in a complex disease such as HD. We have shown how the analysis of microarray data and the integration of publicly available datasets and literature information enables prioritization of associations, such as proteins and mechanisms, that are likely to be involved in HD. In addition, we were able to focus on mechanisms that are associated with epigenetic regulation that may regulate changes that are part of the disease pathology. We argue that such a methodology can be of great value to the scientific community for narrowing down the amount of possible associations but also providing evidence to support a particular hypothesis.
All workflows are deposited in Zenodo (https://doi.org/10.5281/zenodo.16420133 and https://doi.org/10.5281/zenodo.16419834), and the myExperiment platform (http://www.myexperiment.org/packs/553).
Dataset 1: Gene expression data. This folder contains the gene expression data for the three human brain regions and the three mouse brain regions. doi, 10.5256/f1000research.9703.d17946842
Dataset 2: CpG islands. This folder contains the CpG island information both for human and the mouse data. doi, 10.5256/f1000research.9703.d17946944
Dataset 3: Chromatin states for the anterior caudate. This folder contains the four chromatin state data for the anterior caudate. Active TSS proximal promoter: TssA, bivalent regulatory region: TssBiv, heterochromatin: Het, repressed Polycomb: ReprPC. doi, 10.5256/f1000research.9703.d17947046
Dataset 4: Chromatin states for the prefrontal cortex. This folder contains the four chromatin state data for the prefrontal cortex. Active TSS proximal promoter: TssA, bivalent regulatory region: TssBiv, heterochromatin: Het, repressed Polycomb: ReprPC. doi, 10.5256/f1000research.9703.d17947147
Dataset 5: Unique annotations of genes in each chromatin state with Biological Processes. This file contains the results from the semantic analysis with Biological Processes of the genes that were overlapping with active TSS, bivalent TSS, heterochromatin and repressed polycomb chromatin state. The annotations that we present here are uniquely characterizing each gene list (as resulted from the CPA out of the top 50 annotations). doi, 10.5256/f1000research.9703.d17947249
Dataset 6: Annotations of CpG containing genes and non CpG containing genes. The top 50 annotations characterising each group of genes with CpG islands and without, resulted from the semantic analysis of those two groups of genes with Biological Processes. doi, 10.5256/f1000research.9703.d17947352
Dataset 7: Gene prioritization. Top 100 novel proteins, resulted from the semantic analysis associated with HTT and epigenetics. doi, 10.5256/f1000research.9703.d17947453
Dataset 8: Concept profile analysis (CPA) scores for the two gene lists. This folder contains the CPA scores per gene list (top 100 differentially expressed genes in the caudate nucleus and top 1000 differentially expressed from the same brain region) that were obtained by matching the gene lists against the HTT concept profile. doi, 10.5256/f1000research.9703.d17947554
Dataset 9: Percentile scores. This document presents the percentile score of the prioritized gene list when compared to the frequency distribution of 100 match scores of randomly sampled gene-HTT concept pairs out of the 12,391 genes in our concept profile database. doi, 10.5256/f1000research.9703.d17947655
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)