Keywords
HIV-1, novel, correlates of protection, HIV-1 vaccines, HERV-K HML-2, HERV-K102, foamy macrophages, innate immunity
HIV-1, novel, correlates of protection, HIV-1 vaccines, HERV-K HML-2, HERV-K102, foamy macrophages, innate immunity
The extensive revision now discusses in depth why generally adaptive immune response mechanisms are unlikely to significantly affect HIV-1 acquisition rates, and thus why HIV-1 vaccine prevention trials have had poor outcomes. It also expands the discussion of these outcomes with more citations, and why the proposed correlates of protection might be rejected. The introduction of HERV-K102 as a viral anti-virus response and as part of the innate immune system is strengthened in the abstract and opening comments, and the sections describing HERV-K102 attributes in defending against HIV-1 replication and acquisition are presented in a more logical order. As well newer evidence for HERV-K HML-2 (HERV-K102 is a group member) in playing a role in the control of HIV-1 replication is cited. The revision also calls into question whether VE for female gender as reported for the RV144 trial may or may not have been statistically significant as discrepancies in reporting VEs for all participants versus gender were noted. Hopefully the revision clarifies that HERV-K102 is not an exogenously acquired virus, but an endogenous, foamy-like (protector), retrovirus which works in tandem with HERV-K HML-2 activation to reduce the likelihood of integration of a lentivirus/orthoretrovirus (HIV-1). Just like the old adage to fight fire with fire, here it is suggested HERV-K102 as the only known naturally replication competent HERV-K HML-2 group member, may represent a virus-anti-virus response selected for when adaptive immune mechanisms poorly handled lentivirus exposures following the divergence of chimpanzees from humans. HERV-K102 particle production which generates foamy macrophages, along with HML-2 activation, appears to be a new dimension of human innate immunity inversely correlated with triggering adaptive immune mechanisms, which is hypothesized, partially based on clues and on some initial data, to prevent HIV-1 acquisition.
See the author's detailed response to the review by Patricia E Fast
It has been recently stated that immune correlates of protection for HIV-1 prevention vaccines must be complex and/or reliant on the right combination of multiple types of immune responses as a ‘true’ correlate of protection has not yet been characterized (Kuri-Cervantes et al., 2016; Tomaras & Plotkin, 2017). In particular, the V1V2 IgG response as characterized as a primary correlate in the RV144 trial the latter which showed vaccine efficacy (VE) for reduced risk of HIV-1 acquisition (Haynes et al., 2012), by itself is unlikely to signify an immune correlate of protection that can be used to predict vaccine efficacy (VE) outcomes because it was also present in the VAX003 trial (Perez et al., 2017; Yates et al., 2014) that lacked VE (reviewed in Tomaras & Plotkin, 2017). Other objections that can be made include the observation that in the case-control analysis performed among vaccine recipients as reported by Haynes et al. (2012), the error bars for V1V2 IgG overlapped for the comparison of participants who acquired HIV-1 versus those who did not. Accordingly, it was not surprising that the V1V2 IgG response was not significant in the univariate analysis, although it was marginally so in the multivariate analysis. Technically, to be useful as a predictor for vaccine trial outcomes and vaccine development, neither a causal relationship nor protective mechanism needs to be elucidated (Rolland & Gilbert, 2012) as the biomarker may be a surrogate marker of the ‘true’ correlate of protection (Plotkin & Gilbert, 2012). However, despite the clinical testing of over 200 candidate vaccines since 1986 and also reflecting the fact that immune correlates of protection have not been conclusively identified for HIV-1 exposed seronegative (HESN) populations (Safrit & Koff, 2016), the discovery of ‘true’ correlate(s) of protection would be expected to expedite the achievement of HIV-1 vaccines as well as cures. To help this quest along, there are clues that can be derived from the literature as well as from the existing HIV-1 prevention vaccine trials which allude to the nature of the ‘true’ correlate(s) of protection against HIV-1 acquisition. As a result of this inquiry, a novel and plausible candidate innate immune protection mechanism against HIV-1 acquisition has been identified and will be presented. Unexpectantly and quite remarkably, this response appears to be a virus anti-virus response which may have evolved in chimpanzees during exposures to lentiviruses and subsequently acquired by humans less than 2 million years ago.
In terms of randomized clinical trials for vaccine prevention of HIV-1 acquisition, there have been 6 trials, only 3 of 6 which were determined to be informative for correlates of risk (increased or decreased) (reviewed in Stephenson et al., 2016; Tomaras & Plotkin, 2017). They were the HVTN 502 STEP trial (Buchbinder et al., 2008) with follow-up data in HVTN 504 (Duerr et al., 2012), the HVTN 503 Phambili trial (Gray et al., 2011; Gray et al., 2014) with follow-up data in Phambili HVTN 503-S (Moodie et al., 2015) and the RV144 trial (Rerks-Ngarm et al., 2009).
What these three trials had in common was that they all employed viral vectors for the priming/immunization whereas 2 others (Vax004 and Vax003) used proteins with an alum adjuvant and one (HTVN 505) used circular plasmids for priming. Even though the latter HVTN 505 trial used an Ad5 viral vector, it was only used for the boosting (Hammer et al., 2013) and no vaccine efficacy was found. In these informative trials, waning of negative or positive VE (reviewed in Stephenson et al., 2016; Tomaras & Plotkin, 2017) might imply the effects on VE in part related to non-specific effects attributable to use of viral vectors. In addition, in the RV144 trial which uniquely included HIV-1 envelope (Env), evidence of postacquisition selection pressure at 42 months by adaptive immunity may have also contributed to a positive VE (Gartland et al., 2014).
In support of the notion of non-specific effects of the viral vectors employed, Huang et al. (2014) found that while HIV-specific responses measured at 4 weeks after the second vaccination in the STEP trial were not associated with risk of HIV-1 infection, levels of IFN-gamma by ELISpot assay in the mock responses performed in the absence of HIV-1 antigens showed a hazard ratio (HR) of 1.62 (95% CI: 1.28 to 2.04, p<0.001) which was strengthened among participants with pre-existing anti-Ad5 antibodies. Consistent with this finding, IFN-gamma levels are higher in HIV-1 positive over negative individuals (Yong et al., 2016) and are lower in HESN than HIV-1 unexposed controls (Jaumdally et al., 2017). This raises the possibility that induction of innate rather than adaptive immunity by viral vectors may play a more significant role in protection/risk against HIV-1 acquisition than what is currently appreciated. Unfortunately, in none of the informative clinical trials was there a placebo arm which included an empty vector as a control for HIV-1 non-specific or innate immunity effects.
Problematic issues confronting clinical trial interpretation of outcomes include the concern that disparate statistical significance can be demonstrated depending on the statistical and analytical methods employed. This is why it is essential to decide statistical and analytical methods in advance to avoid bias. For the interpretative meaning of finding correlates of risk in randomized, HIV-1 prevention clinical trials which show vaccine efficacy (VE), as alluded to above, the association does not necessarily mean that there was protection against acquisition, as it could also reflect post-acquisition control of replication of HIV-1. For example, while the RV144 trial showed 31. 2% vaccine efficacy at 42 months (Rerks-Ngarm et al., 2009), vaccine efficacy (VE) was 54% versus 3% (p=0.05) in HLA A*02 individuals versus participants lacking this Class I HLA allele in this subgroup analysis. Moreover, VE increased in the HLA A*02 individuals (74% versus 15%, p=0.02) when the subgroup analysis was restricted to acquired HIV-1 strains bearing the lysine residue at site 169 in the V2 domain of the immunizing scaffold epitope (Gartland et al., 2014). However, these authors suggested that this enhanced VE related to postacquisition selection pressure by adaptive immune mechanisms rather than a genuine blocking of HIV-1 acquisition. Nevertheless, this was the first time an HLA allele was associated with VE against HIV-1 acquisition.
Indeed, genome wide association studies have only established CCR5 and certain HLA alleles as being associated with protection against HIV-1 replication and/or acquisition (reviewed in Naranbhai & Carrington, 2017). These alleles, such as HLA-B*27 are also associated with protection against hepatitis C and curiously also associate with autoimmunity (Crux & Elahis, 2017; Khan, 2017). The finding that natural killer alleles appear to interact with HLA-B*57 in control of HIV-1 replication (Tallon et al., 2014), may argue that the HLA-B supertypes which confer protection against HIV or HCV may be through innate mechanisms. In the STEP trial, analysis of host genetics associations identified that the protector HLA alleles (B*27, B*57, B*58:01) known to be associated with HIV-1 control had a lower viral load (Fitzgerald et al., 2011) and showed robust CD8+ T cell activity (Migueles et al., 2011). However, the HIV-1 specific T cell responses measured in the trial did not correspond with the sieve findings on the HIV-1 strains that were acquired and were not associated with lower risk of infection in the vaccine recipients (reviewed in Tomaras & Plotkin, 2017).
The above discussion raises the important but thorny issue of whether or not adaptive immunity per se to HIV-1 specific antigens can actually prevent HIV-1 acquisition. The premise that adaptive immunity can prevent infection which has been clearly shown for a wide variety of pathogens and resulted in successful vaccines, may not be valid for HIV-1. This is because HIV-1 strains show hypervariability in protein sequences, antigen drift and are vulnerable to pseudotyping of its particles by envelope derived from HERV-K retroviruses particularly certain HERV-K HML-2 group members as will be discussed later. Thus, in theory, preformed recognition of HIV-1 specific envelope (Env) epitopes proffered by vaccines whether by antibody or T cell receptors might be expected to only provide postacquisition selection pressure and/or weak correlates of protection. For example, in open label studies involving anti-retroviral treatment interruption, administration of VRC01, a broadly neutralizing antibody targeting the HIV-1 CD4 –binding site found in Env, was shown to significantly delay viral rebound in many persons with HIV-1 infection as measured at 4 weeks when compared with historical controls, but the difference was not significant at week 8 (Bar et al., 2016). HIV-1 is also more complicated than many pathogens as it targets activated immune cells which are generated during immunization. Moreover, adaptive cytokines like IL-1 and IL-2 enhance HIV-1 replication in contrast to the innate cytokines interferon alpha and beta which instead inhibit HIV-1 replication (reviewed in Naif, 2013). These considerations question the fundamental basis for the conduct of prevention HIV-1 vaccine efficacy trials that have the goals of stimulating adaptive immunity.
As discussed by Haynes et al. (2012), no direct evidence in humans was offered for suspecting adaptive immunity may be a strong correlate of protection in preventing HIV-1 infection; only that adaptive immunity modulated progression (postacquisition selection pressure) in humans. In fact, accumulating evidence suggests lowering adaptive responses but without overt immunosuppression may be key to preventing HIV-1 acquisition as determined in HIV-1 exposed seronegative (HESN) cohorts (reviewed in Lajoie et al., 2017). Indeed, the presence of adaptive immunity to HIV-1 antigens does not alter superinfection in high risk individuals (Ronen et al., 2017) providing direct evidence in humans that adaptive responses may not significantly alter HIV-1 acquisition rates. The strongest data taken as a basis for conducting randomized clinical trials to generate adaptive immunity for the prevention of HIV-1 acquisition were derived instead from studies conducted in simians with SIV or SHIV (Haynes et al., 2012). As will be detailed below, a proposed candidate ‘true’ correlate of protection based on innate immunity is notably absent from these models implying simian (macaques) models might not necessarily be predictive for human correlates of risk/protection for HIV-1 prevention vaccines.
The RV144 trial (Rerks-Ngarm et al., 2009) was the only trial that showed vaccine efficacy for prevention of HIV-1 acquisition (reviewed in Haynes et al., 2012; Kim et al., 2015; Kuri-Cervantes et al., 2016; McMichael & Haynes, 2012; Perez et al., 2017; Stephenson et al., 2016; Tomaras & Plotkin, 2017). For this trial conducted in Thailand in heterosexual low risk individuals, the VE of about 60% at one year (6 months after the last immunization) fell to 31.2% at 42 months (Rerks-Ngarm et al., 2009). Although vaccination did not have a significant effect on viral load (p=0.24), the placebo group had 1.26 times better control of viral load over vaccines. This may be consistent with the notion that vaccination might have increased the number of targets, and thereby enhanced HIV-1 replication upon HIV-1 acquisition.
When broken down by gender, females (38.6% VE) had about a 1.5 better VE over males (25.8% VE) in the modified intention-to treat (MITT) analyses, but where the p values were not provided. The provided 95% confidence integrals at -8.6 to 65.3% for females and -7.3 to 53% for males in Table 2 on VE appeared to be anomalous given that in total, the VE in Table 2 was given as 31.2% with 95% CI of 1.7 to 51.8% (ie., if the two components have a negative lower CI how is it possible that the total has a positive lower boundary of CI and the upper boundary lower than the components). Moreover the numbers in Table 2 (MITT) for the calculation of VE (n=15,948) did not match that reported in the abstract for MITT, which instead indicated n was 16,395 and the VE was 31.2% with 95% CI, 1.1 to 52.1; (p=0.04). A quick analysis revealed there were relatively more men removed from the analysis in Table 2 when the MITT based VE for the total population was calculated. The supplemental information did not discuss these discrepancies. Accordingly it is possible but not for certain that females exhibited up to a 1.5 better VE over men in the RV144 trial suggesting a possible female hormone influence on the correlate of risk reduction. The rates of HIV-1 acquisition for females were 0.185 per person-year (pp-y) in the vaccine arm and 0.301 pp-y in the placebo. The corresponding rates for males were 0.197 pp-y in the vaccine arm and 0.266 pp-y in the placebo arm.
With respect to female hormones exhibiting an influence on HIV-1 susceptibility or VE, it should be remembered that in each trial which only enrolled women of child-bearing ages but who were not pregnant nor breast-feeding, women were counseled to practice effective contraception (RV144, Rerks-Ngarm et al., 2009) or required to use hormonal contraceptives and a barrier method (Phambilli HVTN 503, Gray et al., 2011) during the immunization period and for one to several months thereafter. In the STEP trial, no restrictions applied (Buchbinder et al., 2008). In a recent meta-analysis, while the use of various progestins for oral contraception were associated with a significantly increased adjusted hazard ratio of HIV-1 acquisition over women who did not use contraceptives, the combined oral contraceptive was not (Morrison et al., 2015). Thus, a potential confounder of trials involving women is the type and timing of steroid contraception used which should be taken into consideration at the time of trial design, used as a parameter to validate trial randomization, and for checking bias at later time points of analyses. The other two trials which used the same Ad5 vector were stopped early due to an increased risk of HIV-1 acquisition in males discovered in the interim analysis of the STEP trial (Buchbinder et al., 2008). In the extended analysis, in the STEP trial (HVTN 504), there were 172 HIV-1 infections in 1836 males providing a follow-up HR of 1.40 (CI: 1.03 to 1.92, p=0.03) but where there were too few infections in females for HR analysis. The impact of the baseline Ad5 antibodies or lack of circumcision on VE was present in the STEP trial only during the first 18 months (Duerr et al., 2012). In the follow-up for the Phambilli recall trial (HVTN 503-S) with an additional 1286 person-years of follow-up, the total HR was 1.52 (95% CI 1.08 to 2.15, p=0.02). Interestingly in males, the HR was 2.75 (95% CI 1.49 to 5.06, p=0.001) whereas for females it was 1.12 (95% CI 0.73 to 1.72, p=0.62) and non-significant. This trial did not identify Ad5 pre-existing immunity issues possibly due to high seroprevalence at 81%, and similarly did not find circumcision to have any effects in contrast to the STEP trial which focused on high risk men having sex with men. They concluded that significant predictors of HIV-1 acquisition were vaccine treatment, male gender and baseline HSV-2 status (Moodie et al., 2015). Thus of the two trials which were informative for gender effects, females may have exhibited less risk of HIV-1 acquisition related to vaccination or tended towards having higher VE although the discrepancies in the reported analyses of RV144 may need clarification. Taken together with the aforementioned meta-analysis, the issue is raised that estrogen plus progesterone but not progestins alone, might enhance a protector effect against HIV-1 acquisition promoted by viral vectors, which may warrant further investigation.
A number of groups have suggested partial correlates of risk reduction as demonstrated from the RV144 HIV prevention trial, particularly, V1-V2-specific, polyfunctional IgG antibody responses (reviewed in Kim et al., 2015; McMichael & Haynes, 2012; Rolland et al., 2012; Stephenson et al., 2016).
In the RV144 initial analysis, a case-control analysis on samples collected two weeks following the last immunization, it was designed to identify factors predicting HIV-1 infection through 42 months of follow-up in the vaccinated group. It compared 41 vaccinees who became infected with 205 uninfected vaccinees and involved 6 preselected antibody and T cell tests (Haynes et al., 2012). However, since the comparison involved vaccination, there was considerable overlap in the box plots meaning a significant proportion of both those infected or not had the factor being assayed and frequently at the same concentration. This suggested a priori that such a factor may not be a strong candidate for a true correlate of protection. It is unclear if analyses of samples taken 2 to 4 weeks prior to HIV-1 acquisition would have been more informative but this would have presented logistical issues including increased labor and costs for collecting monthly samples.
As reported in the initial study of the potential correlates of protection for the RV144 vaccine trial, it was suggested that there was no role of IgG avidity, ADCC, neutralizing antibody or Env-specific CD4 + T cells in the protective response detected (Haynes et al., 2012). On the other hand, while IgA antibody binding to HIV-1 envelope (Env) directly correlated with acquisition risk in both univariate and multivariate analyses, scaffolded V1-V2 Env IgG antibodies while marginally significant in multivariate analysis, failed to achieve significance in univariate analysis for correlation of protection against HIV-1 acquisition (Haynes et al., 2012). However, secondary analysis suggested that Env-specific IgA antibodies may have interfered with the effects of the protective IgG V1-V2 antibodies.
These findings were subsequently confirmed in different laboratories with different assays and reagents (Zolla-Pazner et al., 2014). Follow-up studies suggested the pertinent Env V1V2 antibodies were IgG3 (Yates et al., 2014), or dependent upon complement activation within the IgG V1V2 population of antibodies (Perez et al., 2017). In the latter case the response rate and magnitudes were higher in RV144 than in VAX003 and VAX004.
In the study by Yates et al. (2014), the IgG or IgG3 response rates (i.e., number of participants with the factor) to the mutated V169K V1V2 strain significantly dropped from testing at 26 weeks to 52 weeks (97 to 11% for IgG and 50 to 3% for IgG3) and the authors suggested a correlation with VE waning shown for the RV144 trial. However, at 52 weeks the VE was about 60% at a time when these antibody responses reached background which instead implied there was no temporal correlation with VE waning. As well, the response rates were not significantly different when compared with visit 5 VAX003 participants where VE was not shown. Moreover, the magnitude of the response was higher in VAX003 visit 5 vaccine recipients for the same epitope. The IgG3 response rates to any of the V1V2 strains tested did not correlate with reduced HIV-1 acquisition except when tested on a V169K mutated V1V2 strain epitope which was significant in both univariate and IgA adjusted analysis. On the other hand, the magnitude of the response to this epitope did not significantly correlate with a reduced risk with or without adjustment for the IgA interference. Interestingly, the response magnitude did reach significance when adjusted for the influence of IgA interference for a number of V1V2 strains but not for the mutated V169K epitope. Taken all together, it seems these findings would not support the contention that IgG3 antibodies induced to V1V2 Env epitopes through vaccination mediated the protection observed.
It should be noted as reviewed in Corey et al. (2015), that the prevalence of K169 in the HIV-1 Env V1V2 region is about 85% in Thailand, while in the RV144 clinical trial, the HIV-1 strains acquired in the placebo-treated group were 83% with K169, while in the vaccine treated it was 66%. This showed that the strain that emerged in the vaccine arm did not match the vaccine, as would be expected. Indeed Rolland et al. (2012) estimated that the vaccine efficacy against viruses matching the vaccine at position K169 was 48% (p value of 0.0036; 95% CI 18–60%) while the vaccine efficacy for mismatched was not significant. This latter finding contradicts the finding of a correlation of the response rate for a mismatched V1V2 epitope, namely the V169K mutation with protection in the RV144 vaccine trial as studied and reported by Yates et al. (2014).
For a more intensive discussion of the various risk and protection factors discovered in HIV-1 vaccine prevention trials, the reader is referred to more comprehensive reviews (Stephenson et al., 2016; Tomaras & Plotkin, 2017).
The relatively low rates of acquisition of HIV-1 per exposure at less than 1 in 1000 for heterosexual transmission (Becerra et al., 2016), might argue that defense against HIV-1 in humans possibly involves a simpler and more potent mechanism than what has yet been elucidated or appreciated. Indeed, that a new scientific paradigm may be needed to advance the development of the HIV-1 vaccine has been proclaimed (Esparza, 2015). What then can we currently deduce about the characteristics of this unknown correlate of risk/protection, taking also into account the results in the three informative HIV-1 prevention vaccine trials?
In any immune response, innate and/or adaptive, activated macrophages control the response. Accordingly, it follows that any risk or protection associated with HIV-1 vaccines must then relate to a key and so far, ill-defined macrophage activation pathway. Moreover, in HIV-1 acquisition, the transmitting/founder strains are generally CCR5-tropic and target macrophages (reviewed in Borggren & Jansson, 2015) as originally shown as early as 1986 (Gartner et al., 1986). Together these findings point to the likelihood that there exists a novel macrophage-based ‘gate-keeper’ defense mechanism that itself determines whether HIV-1 will be acquired, although a role of vaginal myeloid dendritic cells in capturing and disseminating HIV-1 may also play a role in the gatekeeping function (Shen et al., 2014).
The Ad5 vector used in the STEP trial, highly targets liver Kupffer cells, which represent about 80–90% of the macrophages in the body (Khare et al., 2011). Kupffer cells are infected by HIV-1 (Mosoian et al., 2017). In the STEP trial, male uncircumcised participants with Ad5 antibodies were significantly at increased risk of transmission compared with those without vector antibodies (Buchbinder et al., 2008). In contrast, no increased risk of HIV-1 transmission was found in the placebo arm for participants with Ad 5 antibodies. This suggests that at the interface of the Ad5 vector with macrophages, the presence of bound Ad5 antibodies enriched in the local milieu may have somehow blocked the induction of the putative, gatekeeper/defense mechanism of macrophages. With time the levels of the Ad5 vector would have cleared accounting for the waning observed and also since Ad5 seropositivity had no effects in the placebo arm (Duerr et al., 2012). A possible candidate mechanism for this interference by antibody is tuftsin. Tuftsin is a short peptide consisting of the sequence TKPR and is released from bound IgG. Tuftsin is known to inhibit macrophage activation at higher concentrations, while at lower levels, it augments macrophage activation (Siemion & Kluczyk, 1999). Thus, it may be plausible that higher risk participants (uncircumcised men) with pre-existing Ad5 antibodies injected with an Ad5 vector could have experienced inhibition of the putative, novel macrophage defense mechanism, through the local generation of tuftsin, thereby explaining their increased risk. Clearly, this conjecture may warrant further investigation.
Another important clue relates to gender. As discussed above, in the STEP trial (Buchbinder et al., 2008), there were insufficient female participants and HIV-1 infections to address vaccine efficacy in comparison to men. In a follow-up study of the Phambili trial (Moodie et al., 2015), males continued to show heightened risk related to vaccination but which was not found for females. The hazard ratio for men was 2.75 (p=0.001) and for women it was not significant 1.12 (p=0.62) despite high rates of HIV-1 acquisition which implied there was sufficient power for the statistical assessment. In contrast, in the RV144 trial, vaccine induced protection against HIV-1 acquisition was more evident in females than males (about 1.5-fold better) (Rerks-Ngarm et al., 2009), but as mentioned, the statistical significance values reported need re-examination and clarification as discrepancies were noted. All together these findings support the notion that the novel, macrophage based protection mechanism induced by viral vectored vaccines might be enhanced by female hormones.
That the risk enhancement/reduction was only temporary in either trial, generally lasting 6 to 12 months after the last immunization (Buchbinder et al., 2008; Rerks-Ngarm et al., 2009), was consistent with an innate rather than adaptive immunity mechanism.
Since both risk enhancement (Buchbinder et al., 2008) and risk reduction (Rerks-Ngarm et al., 2009) were found with viral vectored vaccines, and not in the VAX003 and VAX004 HIV-1 vaccine trials, which instead involved proteins and adjuvants (reviewed in Shin, 2016), this raises the likelihood that viruses in general may preferentially activate the macrophage-based defense mechanism and/or that adjuvants diminish it.
Overall, these observations point to the existence of a novel, potent innate HIV-1 protection mechanism induced by viruses and enhanced with female hormones, which is launched by alternatively activated macrophages, and that this activation may be sensitive to inhibition by locally bound antibodies, for example as might be the case through tuftsin. Moreover, despite a considerable effort both inside and outside of the trials, convincing immune correlates of risk or prevention against HIV-1 have yet to be revealed. This failure raises the following notions about the mechanism. It is likely not addressed by traditional in vitro culture methods, not discoverable using conventional detection methods on participant samples, such as by examining plasma RNA, microarray and genome wide association studies, nor is it likely present in the rhesus macaque. The critical question becomes, has such a novel, innate, potent, defense mechanism unique to human activated macrophages and difficult to study under standard conditions, been previously described?
Surprisingly, the answer to this may be, yes.
A novel, innate, viral anti-viral defense mechanism unique to humans, associated with the production of foamy macrophages (Figure 1) was serendipitously discovered by scientists working at the Public Health Agency of Canada about 10 years ago, and fulfills all the above criteria for a novel defense mechanism launched by alternatively activated macrophages in response to viruses (Laderoute et al., 2007; Laderoute et al., 2015; Laderoute, 2015). The term alternatively activated macrophages refers to the generation of foamy macrophages which is not observed within studies of adaptive immunity. Moreover, under permissive conditions (culture of cord blood mononuclear cells in IMDM media rather than RPMI) the addition of PHA and IL-2 (various protocols) completely blocked HERV-K102 replication, particle production and the generation of foamy macrophages implying foamy macrophage generation is an alternative to adaptive immunity. HERV-K102 is a non-pathogenic retrovirus with foamy retroviral properties whereby particles accumulate in vacuoles (Laderoute et al., 2015). As expected for foamy retroviruses, capsid assembly occurs in the cytoplasm adjacent to vacuoles (Hütter et al., 2013, and see Figure 1 blue arrow). In these foamy macrophages, HERV-K102 particles are not released through the cell surface and release is instead dependent upon lysis of the foamy macrophages (Laderoute et al., 2015). It is the only human specific HERV-K HML-2 group member (Subramanian et al., 2011) shown to be naturally replication competent in vitro and in vivo (Laderoute et al., 2007; Laderoute et al., 2015).

Left panel, H and E stain of day 11 cultured CB prepared by cytospin. Right panels, electron micrographs show vacuoles in the foamy macrophages contain large numbers of immature particles with envelope spikes. Blue arrow points to the cytoplasmic capsid assembly outside of the vacuole, typical of foamy retroviruses (Hütter et al., 2013). Left panel reproduced under a CC BY- NC 4.0 license (Laderoute et al., 2015). Right panels reproduced with permission from the AIDS journal (Laderoute et al., 2007).
HERV-K102 activation was demonstrated in about 96% of HIV-1 patients which includes particles (75.7%) and/or HERV-K102 surface unit envelope specific antibodies (70 to 80%) (post hoc analysis, Laderoute et al., 2007).
HERV-K102 particles have cDNA genomes rather than RNA due to its foamy retrovirus properties and reversed life cycle to orthoretroviruses (Laderoute et al., 2007; Laderoute et al., 2015). It can be estimated from previously reported data that in HIV-1 patients, there may only be on average about 8,200 HERV-K102 pol cDNA containing particles per ml of plasma with a response rate of about 72% (post hoc analysis of Laderoute et al., 2007). This coincides with estimates of about 8,300 HERV-K HML-2 DNA (transmembrane) env containing particles with a response rate of about 73% from the data of Bhardwaj et al. (2014). It should be noted that two groups have reported the absence of HERV-K HML-2 RNA containing particles in plasma samples from HIV-1 patients (Bhardwaj et al., 2014; Karamitros et al., 2016), suggesting earlier reports of HML-2 RNA in plasma from HIV-1 patients likely were due to cellular contamination. Taken together these results might suggest the only HML-2 particles in HIV-1 patients might be HERV-K102 particles with cDNA genomes.
Several research groups have confirmed that HERV-K102, is induced by HIV-1 in vitro (Brinzevich et al., 2014; Vincendeau et al., 2015). Moreover, HERV-K102 may be the only full length, human specific, HML-2 element induced by HIV-1 and/or Tat (Gonzalez-Hernandez et al., 2012). While the envelope of HERV-K18 and a consensus sequence for HERV-K HML-2 were able to pseudotype HIV-1 virions, interestingly HERV-K102 Env did not (Brinzevich et al., 2014; Lee & Bieniasz, 2007). That HIV-1 may be pseudotyped by HML-2 envelope raises the notion that such pseudotyped particles could help explain, in part, the altered tropism for macrophages bearing the CCR5 coreceptor, which is commonly used by transmitting/founder strains (Borggren & Jansson, 2015). If this is indeed the case, it would also help explain why vaccination against HIV-1 envelope generally fails to prevent HIV-1 acquisition (reviewed in Shin, 2016), or why passive immunization with HIV-1 envelope specific, broadly neutralizing envelope antibodies failed to significantly control viremia upon antiretroviral treatment interruption (Bar et al., 2016; Caskey et al., 2017).
In potential substantiation of an important role of HERV-K HML-2 in the control of HIV-1 replication, HERV-K HML-2 gag and envelope RNA expression in peripheral blood mononuclear cells (PBMCs) in HIV-1 patients were shown to be inversely correlated with T cell activation markers (Ormsby et al., 2012). Since it is known that activated T cells correlate with HIV-1 progression (Deeks et al., 2004), this implies HML-2 expression generally, and by proxy HERV-K102 activation, may antagonize HIV-1 replication and/or the HML-2 response is an alternative to adaptive immunity. Indeed, recent evidence suggests that the phylogenetically newer HERV-K HML-2 elements containing LTR5Hs (which include HERV-K102) were upregulated in CD11c+ myeloid dendritic cells isolated from HIV-1 patients, whereas, in normal healthy controls, the older LTR5A and LTR5B bearing HML-2 elements prevailed (Young et al., 2014). Thus, the HML-2 response to HIV-1 involves primarily human specific HML-2 group members which would and does involve HERV-K102 (Laderoute et al., 2007).
Both antibodies and T cell responses to HERV-K HML-2 and/or HERV-K102 envelope have been demonstrated in HIV-1 and breast cancer patients (reviewed in Laderoute et al., 2015). A T cell clone isolated from an elite controller, which recognized a peptide identical to HERV-K102 envelope, was shown in vitro to specifically eliminate cells infected with various HIV and SIV strains (Jones et al., 2012). More recent evidence from the same group showed elite controllers have stronger antibody and T cell responses (rates and magnitude) to HERV-K HML-2 gag than viremic controllers and normal healthy controls (de Mulder et al., 2017). Altogether these results imply HERV-K HML-2 activation which includes HERV-K102 particle production (Laderoute et al., 2007), may play an important role in controlling HIV-1 replication.
Remarkably, a monoclonal antibody made to HERV-K102 surface unit of envelope could directly provoke apoptosis in vitro and in vivo of breast cancer cells (Wang-Johanning et al., 2012). This might suggest that the expression of HML-2 envelope on the surface of virally infected or transformed cells, but which is not found on normal cells, plays a more active role in innate host protection than merely as a surrogate marker. These findings may also further document the unexpected potency of this innate protector mechanism against HIV-1, which unlike adaptive immunity, functions irrespective of the hypervariability of HIV-1, quasi-species and/or strains of HIV-1 or even lentivirus involved (Jones et al., 2012).
In preliminary experiments, increased HERV-K102 integration (mean about 5 fold) over normal healthy controls was associated with protection against HIV-1 acquisition in a female, HIV-1 highly exposed seronegative cohort (HESN), at the level of about 80% of the tested cohort which was not detected in HIV-1 infected individuals irrespective of their use of anti-virals (Laderoute et al., 2015). This is consistent with high integration levels reported for foamy viruses in hematopoietic cell lines in vitro (Meiering et al., 2000). It should be noted that the analysis was performed on genomic DNA isolated from plasma (where cDNA was digested to avoid interference) which in all likelihood may have favored analysis of recently lysed cells, such as macrophages releasing HERV-K102 particles. Increased integration of HERV-K102 was also found in vitro associated with HERV-K102 replication (Laderoute et al., 2015).
It should also be noted that increased integration in vivo provides direct evidence that HERV-K102 particles are infectious. Dube et al. (2014) have shown with a resuscitated HERV-K HML-2 virus construct, that both viruses with DNA and RNA genomes are infectious.
Moreover, protection in the infamous Nairobi HESN cohort (Fowke et al., 1996) is known to be temporary, as resistance to HIV-1 acquisition dwindled as early as 6 months to a year following abstinence from sex trade work (Kaul et al., 2001). Thus, HERV-K102 which might be implicated in the Nairobi HESN cohort, potentially, may meet a key criterion of temporary activity and waning after 6 to 12 months. This would also be expected for innate immune mechanisms more generally.
HERV-K102 is a member of the HERV-K human specific HML-2 group and has hallmark features of foamy viruses (Laderoute et al., 2015). This raises the notion that HERV-K102 may be a candidate foamy virus of humans which has alluded investigators. Foamy viruses are unconventional retroviruses with a reversed life cycle to orthoretroviruses (Linial, 1999; Rethwilm & Bodem, 2013). They are not pathogenic and co-speciated in primates over the past 30 years (Switzer et al., 2005). Accordingly, foamy viruses likely serve a beneficial role to the host. In this regard, accumulating phylogenetic evidence is consistent with a potential role of HERV-K HML-2 in limiting invasion by orthoretroviruses (Magiorkinis et al., 2015). Ancestral HML-2 elements emerged about 10.3 million years ago (Mya) (Subramanian et al., 2011). There has been a striking decline of insertions of ERVs in the last 10 My in the genomes of all sequenced hominids (great apes and gibbons), but not in old world monkeys (baboons and macaques), particularly regarding HERV-H (Magiorkinis et al., 2015). HERV-H makes up 88% of all the ERV integrations into the human genome in the last 30 My and became extinct over the past 10 My. HERV-H is a gammaretrovirus, which integrated around 45 to 60 Mya and has about 962 copies in the human genome (Chuong et al., 2016). HERV-K, with 10 groups in the clade, only one of which is HML-2, on the other hand, entered the genome of ancestral catarrhines about 32 to 44 Mya, after the split from New World monkeys and before the split of hominids from the Old World monkeys (Kim & Han, 2015). The sister lineages of HERV-K in most other catarrhines appear to have become extinct. Most remarkably, the HERV-K HML-2 group in humans is the only HERV-K that has continued to replicate since the origin of the catarrhines (Magiorkinis et al., 2015). HERV-K102 is a member of the bioactive HERV-K HML-2 group and appears to be the only known replication competent member both in vitro and in vivo (Laderoute et al., 2007; Laderoute et al., 2015).
Accordingly, since phylogenetic evidence supports an association of HERV-K HML-2 activity with protection against integration of orthoretroviruses (i.e., acquisition), this may help substantiate the notion that modern day HERV-K102 particles, along with expression of proteins from other HML-2 elements, might prevent HIV-1 acquisition.
Somewhat ironically, humans apparently acquired the HERV-K102 defense mechanism from the same source of the modern HIV-1 pandemic strain; namely, chimpanzees, possibly between 500,000 and up to 2 Mya (Romano et al., 2006; Subramanian et al., 2011).
The Homo-Pan split has been estimated at 6.6 Mya (Magiorkinis et al., 2015) or earlier at 7-8 Mya (Langergraber et al., 2012). As mentioned, the HERV-K HML-2 elements originated in primates about 10.3 Mya and the CERV-K102 sequence (DQ112149), which is 97% identical to HERV-K102, was estimated to have integrated into chimpanzees at a non-orthologous position about 10 (+/- 3.3) Mya (Romano et al., 2006). Lentiviruses may have been active in primates since the divergence of chimpanzees and humans (Katzourakis et al., 2007; Sawyer et al., 2004). Moreover, it has been suggested the ancestor to HIV-1 may have arisen in chimpanzees about 4 Mya (Gifford, 2012). Since it has been reported that subsets of chimpanzees with chronic HIV-1 infection showed progression analogous to humans, including greater expression of CD38 in CD8+ HLA-DR+ T cells (O’Neil et al., 2000), this raises the notion that an HERV-K102 ancestor, as a potential antidote for HIV-1 infection may have been selected through evolution in chimpanzees before it was acquired by humans. Accordingly, it is possible over about a 2 million-year window or longer, the HERV-K102 ancestor may have adapted to HIV-1 like ancestor lentiviruses in chimpanzees prior to its acquisition by humans. Thus, the phylogenetic evidence raises the notion that HERV-K102 as a replication competent HERV-K HML-2 retrovirus, may have evolved to limit HIV/lentivirus replication and genome invasion. Testing of this hypothesis could be done by examining the bioactivity of HERV-K102 and CERV-K102 particles on HIV-1 infected human and chimpanzee cells under in vitro conditions permissive for HERV-K102 versus HIV-1 replication.
When cord blood mononuclear cells (CB) were cultured in IMDM rather than RPMI media, HERV-K102 spontaneously replicated, generating high levels of foamy macrophages (Laderoute et al., 2007; Laderoute et al., 2015). Others have similarly reported the induction of foamy macrophages when CB was cultured in IMDM (Stec et al., 2007). From Stec et al. (2007) the foamy macrophages produced under these conditions were identified as CD14 ++/CD16+, whereas HIV-1 is known to also preferentially infect these macrophages as opposed to other macrophage subpopulations (Ellery et al., 2007). Indeed vacuoles in HIV-1 infected macrophages were previously observed (Gartner et al., 1986). It may be critical to HIV-1 acquisition that HIV-1 first infects macrophages which are the ones which produce HERV-K102 particles to inhibit this protector response.
It is notable that HERV-K102 can be quickly and strongly induced in vivo increasing from no particles detected in plasma to 2.55 × 1011 cDNA containing particles per ml of plasma within 84 hours (unpublished study; Marian Laderoute). This rapid and intense induction is likely to be critical to whether HIV-1 is acquired or not. While high levels of particles at 1010 to 1012 per ml of plasma were frequently found in patients viremic for various bloodborne pathogens, the maximum levels in HIV-1 patients were notably 7 to 8 log-fold downmodulated in comparison (Laderoute et al., 2007). These results are consistent with the notion that upon HIV-1 acquisition, HERV-K102 particle production is strongly inhibited. This contrasts with HESN where 5 fold elevated integrated levels of HERV-K102 pol were demonstrated in genomic DNA isolated from plasma (Laderoute et al., 2015) which was suggestive of previous high levels of particle production.
HERV-K102 particles released by freeze-thaw cycles of cultured CB cells, induced rapid and complete cell lysis of MRC-5 cells at 24 hours, which was not demonstrated for other cell lines tested in parallel (unpublished study; Marian Laderoute). This was expected as foamy viruses are well known to produce rapid cell lysis of some, but interestingly, not all fibroblastic cell lines (Linial, 2001). However, it importantly remains to be determined if HERV-K102 particles might similarly rapidly lyse HIV-1 infected cells.
In terms of other characteristics of the defense mechanism deduced earlier from the informative HIV-1 vaccine clinical trials, combination female steroid hormones (estrogen then progesterone) have been shown to stimulate the expression of HERV-K HML-2 (Ono et al., 1987). As mentioned, in a recent meta-analysis, while the use of various progestins for oral contraception were associated with a significantly increased adjusted hazard ratio of HIV-1 acquisition over women who did not use contraceptives, the combined oral contraceptive was not (Morrison et al., 2015). Thus, HERV-K HML-2 activity may be enhanced by female combination hormones, whether in response to viral vectors, viruses or not.
Relevant to the increased risk of HIV-1 acquisition related to Ad5 antibodies in the STEP trial (Buchbinder et al., 2008), at a high concentration (2 mg/ml), tuftsin inhibited the production of HERV-K102 DNA in cultured cord blood mononuclear cells (CB) by 53%, while at a lower concentration (200 ng/ml), tuftsin enhanced the replication of HERV-K102 pol containing DNA over normal genomic levels by 237% (unpublished study; Marian Laderoute). Thus, it seems as a protector mechanism launched by alternatively activated macrophages, HERV-K102 particle production might be subject to modulation by tuftsin and thus possibly relevant to the adverse outcomes of the STEP trial. Clearly, further investigation of the mechanisms of how pre-existing antibodies were associated with adverse outcomes in the STEP trial appears to be warranted and should include studies on levels of tuftsin.
The identification and elucidation of correlates of protection against HIV-1 have been challenging. Overall the failure to identify HERV-K102 particles pertains largely to the notion that its presence is, more often than not, overlooked or not addressed by standard methodological approaches. For example, because HERV-K102 is unique to humans (Subramanian et al., 2011), it is absent from animal models, such as macaques and rodents, which are commonly used for vaccine or immunological investigations. In addition to HERV-K102 replication being inhibited when PBMCs or CBs are cultured in the more traditional RPMI media invariably used by immunologists (Argaw-Denboba et al., 2017; Laderoute et al., 2015), HERV-K102 activation is also blocked by the depletion of CD14 + cells from PBMC, and also by the addition of PHA and IL-2 to cultures performed in IMDM (unpublished studies; Marian Laderoute). It should be noted that the latter also raises the important notion that adjuvants might inhibit HML-2 activation and HERV-K102 particle production whereas viral vectors may preferentially activate this alternative innate mechanism. Accordingly, it may not be a co-incidence that the conditions that block HERV-K102 particle production in vitro are those that instead are commonly employed to demonstrate HIV-1 infectivity, such as purified T cells activated with PHA and IL-2 cultured in RPMI. Indeed, these observations would be consistent with the possibility that HERV-K102 particles may antagonize HIV-1 replication in vitro. Importantly, while this needs to be directly examined for HERV-K102 particles, interference of HIV-1 replication/infectivity by HML-2 elements has been previously reported (Monde et al., 2012; Padow et al., 2000).
The detection of the presence of HERV-K102 particles also eludes other common approaches utilized for investigations. For example, detection of particles in plasma requires an alternative isolation strategy seldom employed by retrovirologists. It requires DNA and not RNA isolation from plasma (Laderoute et al., 2007), where the common use of DNAse would be contraindicated. As well, genome wide association studies and microarray analysis typically exclude highly repetitious sequences (Baranzini et al., 2010; Held et al., 2003, respectively) to which this element belongs. Accordingly, HERV-K102 particle production appears to have eluded the field due to the difficulty in demonstrating its presence using standard or traditional approaches.
This inquiry has led to the notion that HERV-K102 particle production, which alternatively generates foamy macrophages as part of innate immunity, appears to fulfil the requirements of a deduced candidate correlate of protection against HIV-1 acquisition. HERV-K102 appears to be a ‘virus anti-virus’ response which may have evolved in chimpanzees during exposures to lentiviruses and subsequently acquired by humans less than 2 million years ago. HIV-1 is a challenge for adaptive immunity due to the high levels of antigenic heterogeneity, potential pseudotyping such as by HML-2 Env, that some adaptive cytokines enhance HIV-1 replication, and that HIV-1 infects activated immune target cells. In contrast, the HERV-K102/HML-2 paradigm of defence relies upon recognition of surrogate HML-2 surface markers on HIV-1 infected by HML-2 antibodies or T cells, and that HML-2 elements interfere with HIV-1 infectivity and/or replication. The HERV-K102 virus, in theory, may undergo lytic replication within HIV-1 infected cells where high levels of preformed HERV-K102 particles in the host may potentially block acquisition. Accordingly, it will be very important to study lytic infections of HERV-K102 in HIV-1 infected cells and Tat transformed cells to confirm or disprove this hypothesis.
This candidacy has been strengthened by biological, clinical and phylogenetic evidence, including that which implies preformed HERV-K102 particles may be associated with protection against HIV-1 acquisition. That conversely, acquisition of HIV-1 would be associated with significantly log-lower levels of HERV-K102 particles, would be anticipated and was observed. Given also the preliminary evidence that tuftsin at higher levels could block the replication of HERV-K102 in vitro, suggests the blocking of the same mechanism, such as by Ad5 antibodies in the STEP trial shown in males at higher risk, could plausibly account for the increased risk observed in this informative trial. Finally, that the host source of this remarkable virus anti-virus innate protection mechanism appears to be the same as that for pandemic strains of HIV-1 would strengthen its authenticity, especially given the likelihood of millions of years of co-evolution of the HERV-K102 and the HIV-1 ancestors in chimpanzees. Overall, the available evidence substantiates that a special antagonistic relationship exists between HIV-1 and a foamy-like retrovirus, HERV-K102.
Accordingly, it will be extremely important to prioritize the testing of human endogenous retrovirus K102 (HERV-K102) particle production, integration, and/or envelope specific antibody production to prove or disprove it as a correlate of risk/protection on actual STEP, Phambili and RV144 clinical trial participants (Figure 2). Exploratory studies in other HESN cohorts, in elite controllers, and in post treatment interruption controllers, may also serve to further strengthen or dismiss the correlation. No less significantly, the clinical ramifications of pseudotyping of HIV-1 virions by HML-2 envelope (Brinzevich et al., 2014), needs to be addressed as it may also help explain in part the failed vaccine and cure attempts and possibly co-receptor usage by transmitted/founder HIV-1 strains.
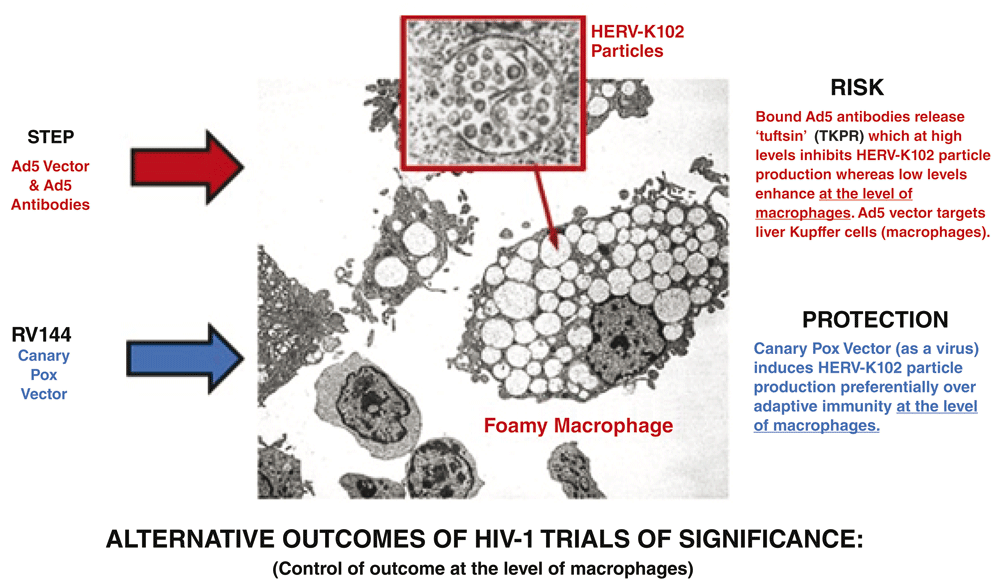
Adapted from Figure 1.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)