Keywords
epitranscriptomics, RNA-seq, RNA editing, differential RNA editing, editing-per-kilobase, EPK
This article is included in the Bioinformatics gateway.
epitranscriptomics, RNA-seq, RNA editing, differential RNA editing, editing-per-kilobase, EPK
In this revised manuscript, a brief description of RNAEditor was added in the Methods section. Furthermore, the required hardware configuration for running DRETools, along with run times when analyzing each testing sample, were added.
See the authors' detailed response to the review by Yicheng Zhao
RNA editing is a class of epitranscriptomic post-transcriptional modification found throughout metazoa consisting of the abundant conversion of adenosine-to-inosine (A-to-I) by ADARs (adenosine deaminases acting on RNA) and rare conversion of cytosine-to-uridine (C-to-U) by APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like)1. RNA editing is particularly interesting as it is detectable as A-to-G and C-to-T mismatches to the reference genome within standard RNA-sequencing data via specialized computational pipelines2. An increasing number of studies link changes in editing at specific sites or clusters-of-sites to diseases, such as epilepsy and atherosclerosis3,4. Yet, no software for detecting differential editing is available. To meet this need, we present DRETools5: 1) to calculate units that help reduce sample-bias, similar to FPKM for RNA expression; and 2) to find differentially edited sites and editing islands (i.e., clusters of editing sites)6. Further, we showcase two examples of finding differential editing and related tasks with DREtools7
DRETools can be run via command-line by typing “dretools”, which will print the main help menu. The main help menu contains a list of operations that are available from dretools with short descriptions of each operation’s purpose. To run an operation, type dretools followed by the operation name. Further detail on each operation, including available command-line arguments and usage examples, can be found by running an operation with the --help argument. On the main help menu, operations are organized into sub-headings based on similar functions. Further detail of each sub-heading and corresponding operations can be found in the following sections.
DRETools requires the output from RNA detection software. Here we used RNAEditor6 to detect editing sites in standard RNA-seq data. RNAEditor uses a specialized alignment and variant calling pipeline to find potential editing sites and then uses filters to remove false positives. In addition to RNAEditor, there are a number of other editing detection tools available. DRETools is usable with any of these tools that work in a similar matter and produces a VCF file containing editing sites and a BAM file containing aligned reads. Details regarding the usage of RNAEditor (e.g. analysis pipelines, configuration files, and non-downloadable reference files) can be found in the archived data5.
One fundamental problem between groups of samples is a lack of standardized units for describing editing within samples, editing islands, and sites. To this end, DRETools implements Editing Per Kilobase (EPK) based on “overall editing” (OE)8. EPK builds upon OE by considering both A-to-G and C-to-T transitions, excludes editing sites with 100% edited bases as potential mutations, and scaling by 103 for readability (similar to FPKM). EPK is calculated by dividing the total number of “edited” bases by the total number of bases overlapping known editing sites and multiplying by 103. In addition to samples, DRETools can compute EPKs for editing islands and sites. Sample-wise editing can be computed with the “sample-epk” function and can be thought of as the global-editing-rate, whereas, the EPK of islands and sites can be computed with "region-epk" and "edsite-epk" respectively, and thought of as the “local-editing-intensity”.
Recently, a method was developed to find differentially edited sites between epileptic or control mouse hippocampi3. However, methods capable of comparing different tissues are also needed. The problem is that unless the global-editing-rates are similar, we cannot determine if changes are due to differing global-editing-rates or other phenomena, such as competition with N6-methyladenosine (m6A)9. Furthermore, ADARs have been described to edit both specific sites in some cases and non-specifically within small regions in other cases10. Therefore, in addition to individual editing sites, looking at the clusters of editing is also of interest. DRETools addresses both these issues by allowing the normalization of both the global-editing-rate and site or island local-editing-intensity in EPK and testing for differential editing using a linear model (LM) with the formula: "logFeatureEPK ~ logSampleEPK + featureLength + averageReadDepth" (features can be sites or islands), which adjusts expectations for what constitutes differential editing.
DRETools also includes various helper functions. For example, the merge section contains functions to find editing islands6 and create consensus sets of editing sites by merging sites from multiple samples. Finally, the stats heading contains functions that calculate useful information about editing at the sample, gene, and site levels, such as the editable area or the number of editing sites falling in 3’/5’-untranslated regions, introns, or exons.
Minimum requirements for DRETools are 8 gigabytes (GB) of RAM, a 100 GB hard drive, and an operating system with a Bash command-line interface, R version 3.3+, and Python version 3.5+. The first two operations required on average (n=5) 4.2 minutes (min) and 449 megabytes of RAM memory (MB) for edsite-merge and 4.4 min and 192 MB for find-islands. The benchmarks of remaining operations are primarily dependent on the BAM files used for computation. The BAM files used here ranged from 3.2-29 GB and 31-282 million reads. Performance was as follows: sample-epk (6-42 min, 40-54 MB), edsite-epk (6-41 min, 40-310 MB), region-epk (7-35 min, 40-323 MB), edsite-diff (0.41-3.49 min and 534.09-2280 MB), and region-diff (0.05-0.23 min, 190.31-522 MB).
To illustrate the utility of DRETools, we surveyed differential editing in human umbilical vein endothelial cells (HUVEC) transfected with either an siRNA against ADAR1 or against a random sequence (control)4 and the immortalized cell lines GM12787 and K56211. First we surveyed sample-wise editing using the function “sample-epk.” (Figure 1A,B). Using EPK reduces variation within groups compared to the usage of number of editing sites. For example, the coefficient of variance drops from 0.21 to 0.05 for the silenced ADAR1 group and 0.52 to 0.01 for the control group. Similarly, when comparing the immortalized cell lines, the coefficient of variance is reduced from 0.57 to 0.25 and 0.46 to 0.11, respectively (Figure 1C, D).
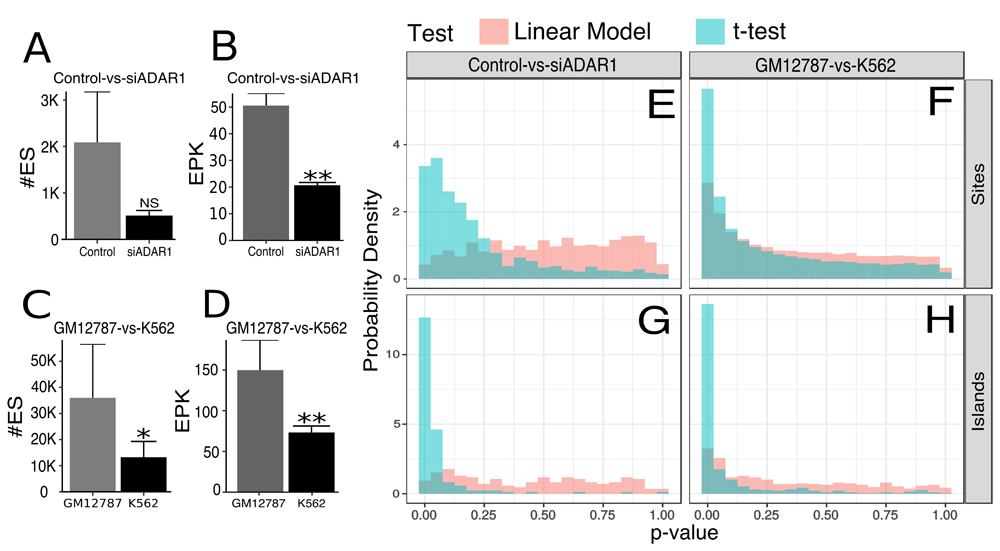
(A) The number of editing sites in HUVEC control and silenced ADAR1 groups (p=0.77). NS, p>0.05. (B) HUVEC control and silenced ADAR1 (siADAR1) represented in EPK (p=0.7.8E-5). **p<0.0001. (C) The number of editing sites detected in GM12787 and K562 cells (p=1.2E-3). *p<0.05. (D) Editing in GM12787 and K562 cells represented in EPK (p=2.5E-6). **p<0.00011E-4. (E–H) Histograms detailing the distribution of p-values when testing for differential editing in a site- or island-wise manner. The site-wise comparison between: (E) siADAR1 and control; and (F) GM12787 and K562 cells. The island-wise comparison between: (G) siADAR1 and control; and (H) GM12787 and K562 cells.
Next, we compared the EPKs of editing islands within the immortalized cell lines using “epk-region”. Using EPK to represent editing islands as opposed to the number of edited bases reduces the coefficient of variance from 0.60 ± 0.21 to 0.31 ± 0.11 (p=2E-30). Finally, we tested for differential editing using the functions “region-diff” for islands and “site-diff” for editing sites (Figure 1E–H). Comparing silenced ADAR1 to the control, the LM yielded a uniform distribution of p-values. In contrast, when using t-test applied to the same data, the distribution of p-values is shifted to the left and exhibits greater skew. However, in the immortalized cell lines, p-values calculated by the LM are more leftward skewed while p-values from the t-test became more uniformly distributed. This provides evidence that the LM can effectively reduce type I errors when testing for differential editing. For example, the LM correctly recognizes that most of the differences between the silenced ADAR1 and control groups arise from the reduction of the global-editing-rate in the silenced samples. Whereas the t-test, which does not consider the global-editing-rates, finds many differentially edited sites and islands. Conversely, when comparing the immortalized cell lines, despite the large difference in EPK, many differentially edited sites and islands are detected. While deeper biological validation is needed to be certain, these could be instances of some other phenomena, such as m6A9, affecting the editing in individual sites or islands.
DRETools is a command-line tool suite for finding differentially edited sites and islands. It allows users to calculate units that reduce sample-bias and find differentially edited sites and islands even when the global-editing-rate of groups being compared is different. Furthermore, it also includes a variety of other features for exploring RNA editing. These make DRETools a valuable tool for further investigating epitranscriptomics.
All RNA-seq data are publically available and were downloaded from the NCBI SRA database12. The HUVEC data sets were generated by Stellos et al., 20164 and the GM12787 and K562 cells by the ENCODE project11. Lists of accession numbers, pipelines used to generate analyses, and intermediate files generated are archived on Zenodo7.
Source code available from: http://dretools.bitbucket.io/.
Data and analysis pipelines: https://zenodo.org/record/14006485.
Source code at time of publication: https://zenodo.org/record/14000057.
License: The software, and data and analysis pipelines are available under a Creative Commons Attribution 4.0 International (CC BY 4.0) license.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)