Keywords
genetic screen, network biology, network propagation, CRISPR
This article is included in the Bioinformatics gateway.
This article is included in the Genomics and Genetics gateway.
This article is included in the GenomeSpace collection.
genetic screen, network biology, network propagation, CRISPR
The rise of next generation sequencing technology and CRISPR gene editing technology has opened up new opportunities for high throughput genetic screens. Increasingly systems biology and molecular networks are becoming more important in the analysis of the mechanisms that are implicated in these screens. Here we present a GenomeSpace recipe providing a standardized pipeline for combining the analysis of a screen with networks and represents a logical next step in providing user-friendly bioinformatics workflows for these types of screens.
This recipe provides a way to process the results of a CRISPR-Cas9 genome wide knockout screen. In such a screen, single guide RNAs (sgRNAs) are designed to target and knock down genes by binding to the target gene and introducing double strand DNA breaks. (Koike-Yusa et al., 2014). Those bound mRNA are subsequently digested by the Cas9 complex and thus do not yield a gene product. In a cell, if the sgRNA is introduced for a gene that is essential for the survival of the cell, that cell will die, and the sgRNA will be depleted. Thus, by sequencing the sgRNAs and looking for a depletion of the sgRNAs targeting a particular gene, we can infer the essentiality of that target gene. Since a large number of sgRNAs can be introduced in a single screen, the essentiality of many genes can be tested at once. However, there are challenges that can arise in the normalization and processing of the read counts; often more than one sgRNA corresponds to a gene but with different efficiency, and significant biases exist in sequencing different sgRNAs. For these reasons the MAGeCK (Li et al., 2014) method was developed to handle data resulting from such a screen.
On the systems biology side of the analysis, we have chosen to employ network propagation as a method of identifying subnetworks representing inferred mechanisms that are implicated by the CRISPR screen. Network propagation has become an essential tool in many network applications; it has been used to identify mechanisms of cancer (Leiserson et al., 2015), to implicate genes in GWAS studies (Qian et al., 2014), and to find functional modules (Vanunu et al., 2010). Network propagation considers genes as nodes on the graph of a biological network. It performs a random walk along the edges of the graph from a set of query nodes. We expect that genes that are implicated in a phenotype will occur in regions of the network that represent mechanisms that are relevant to the screen conditions, and so the random walk will be likely to land on relevant genes. Genes that are near query nodes are therefore implicated by association. For a review of the many flavors and applications of network propagation, see (Cowen et al., 2017).
An overview of the pipeline appears in Figure 1. The recipe begins using the raw read counts of sgRNAs as input to the MAGeCK module in GenePattern (www.genepattern.org). After normalizing the data, MAGeCK detects differential read counts for each sgRNA using an over-dispersed Poisson model. Next, it detects statistical underrepresentation of the sgRNAs corresponding to particular genes to infer that a gene is essential for the survival of a cell. The reasoning behind this is that if a sgRNA targets an essential gene, the cells that contain that sgRNA will not replicate and the sgRNA will be underrepresented compared to other genes.
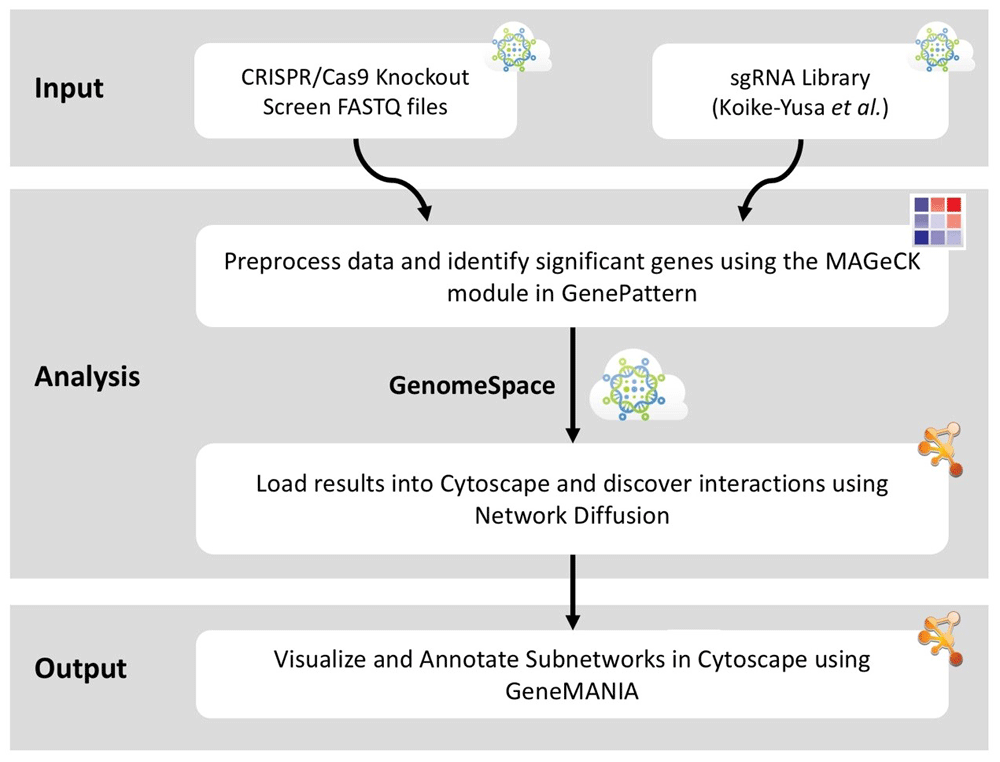
This recipe shows how using GenomeSpace seamlessly integrates multiple bioinformatics tools into a single, easily reproducible pipeline. The publicly available preprocessed knockout screen files and sgRNA library from Koike-Yusa et al. can be transferred directly from GenomeSpace to the ported MAGeCK module in GenePattern (Li et al., 2014). By exporting the resulting list of significant genes to GenomeSpace, the data can be imported directly to Cytoscape without having to download the files locally. Cytoscape’s integrated plugins for NDEx (Pratt et al., 2015), Network Diffusion (Carlin et al., 2017; Cowen et al., 2017), and GeneMANIA (Montojo et al., 2010) allow for the remainder of the recipe to completed within its user-friendly environment.
After we determine a set of essential genes, we pass their identity via GenomeSpace to Cytoscape (Shannon et al., 2003). All analysis in Cytoscape is based on networks, thus a relevant reference molecular network must be imported; the recipe uses the NDEx database (Pratt et al., 2015) to identify such a network. In this case, we choose the National Cancer Institute’s Pathway Interaction Database (Schaefer et al., 2009). The set of essential genes, i.e., the hits from the genetic screen, are imported from GenomeSpace as a table, then used as the seed nodes for network propagation.
The propagation process starts with a single unit of “heat” on each of the nodes that represent the genes that are found to be underrepresented in the screen, and therefore essential for the growth of the cell. We use a heat diffusion process, treating the network as an unweighted, undirected graph. Heat diffusion smooths the original signal over the network, iteratively passing the signal on each node to its neighbor. It identifies regions of the network that have a high concentration of hits. Here we use a time parameter (which represents the amount of time that the heat is allowed to diffuse over the graph) of 0.1. This is a common choice for the time parameter (see Paull et al., 2013). The recipe employs the network diffusion service built into Cytoscape natively (Carlin et al., 2017).
Next, applying a cutoff of the top 200 genes with the most heat after diffusion, we choose a subnetwork that has a high concentration of hits. Finally, in order to understand the composition of the subnetwork, we apply the GeneMANIA Cytoscape plugin. For any network, GeneMANIA (Montojo et al., 2010) shows what functional Gene Ontology categories are enriched in that network.
We used a previously published CRISPR study (Koike-Yusa et al., 2014) to illustrate the use of this pipeline. In this study, the authors use mouse embryonic stem cells grown in the presence of alpha-toxin. This screen was therefore designed to expose the genes involved in the mechanism of resistance to the toxin. The largest connected network component of the top 200 genes after propagation appears in Figure 2.
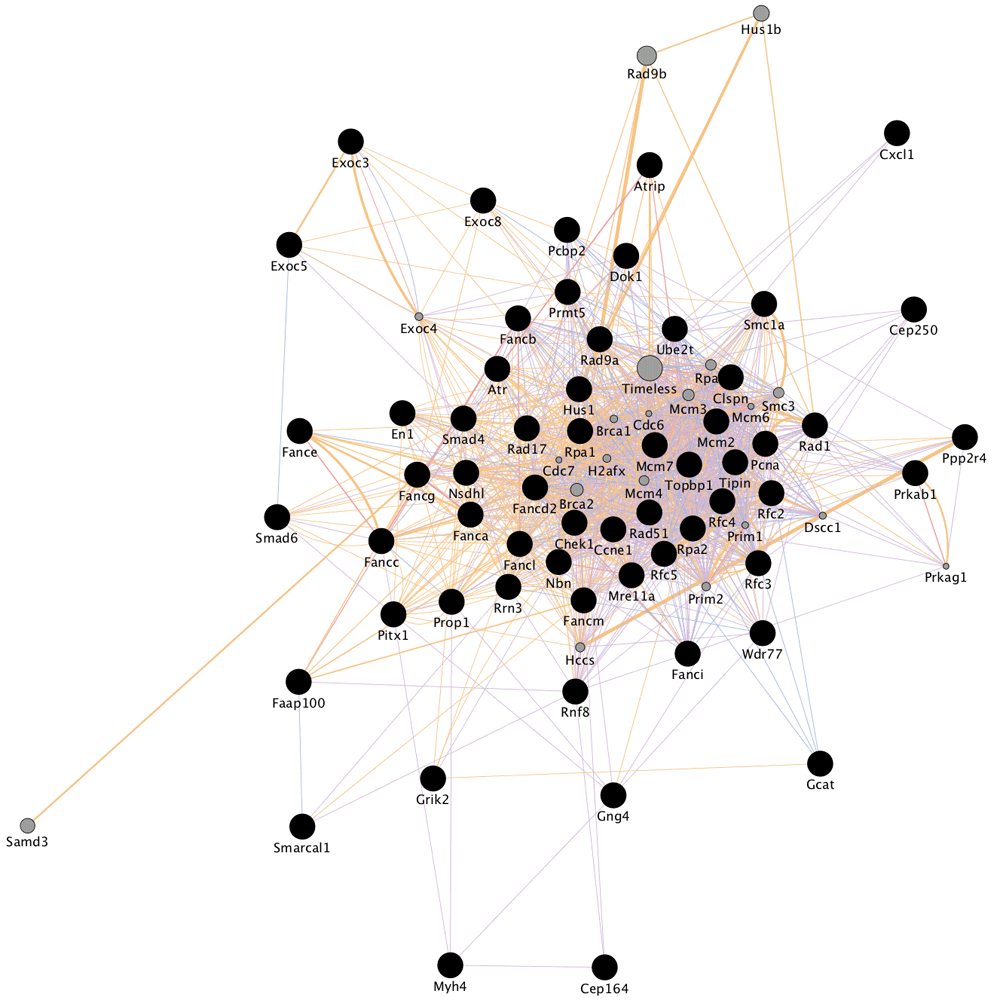
The black nodes represent genes that are significantly deplete in a CRISPR screen. Grey nodes represent genes that are closely associated with the hits by the network and are scaled by the strength of their association.
The results of the GeneMania enrichment suggest that DNA repair is the single most important gene set in handling alpha-toxin. This is consistent with the findings in (Bantel et al., 2001) that alpha-toxin causes an influx of monovalent ions that can cause DNA fragmentation. In the absence of DNA repair machinery, the cells cannot recover from this stress and therefore die. The complete table of the Gene Ontology terms that were significantly enriched in the subnetwork appear in Table S1.
There are several variations that can be used depending on preferences of the user. For example, different tools such as DESeq (Anders & Huber, 2010) and edgeR (Robinson et al., 2010) can be used to identify the hits. Also, the final biological interpretation of the subnetworks was performed with the GeneMania plugin, but the gene list can also be exported and interpreted by another annotation tool. Another approach is to export the gene list and corresponding heats using GenomeSpace and use the Molecular Signature Database gene set overlap tool (http://software.broadinstitute.org/gsea/msigdb/index.jsp), (Liberzon et al., 2011) applied to the genes in the identified subnetwork.
The recipe and Koike-Yusa et al. datasets are publicly available at http://recipes.genomespace.org/view/75. GenomeSpace, an open-source bioinformatics tool, serves as the data highway allowing for seamless transfer of information between tools, and can be found at http://www.genomespace.org/. The MAGeCK algorithm has been wrapped as a GenePattern module, which can be run locally or on the public GenePattern servers at http://genepattern.org. Additionally, GenePattern has added Jupyter Notebook compatibility through GenePattern Notebook (http://genepattern-notebook.org/). Finally, Cytoscape and all associated plugins (ie. GeneMANIA and NDEx) can be found at http://www.cytoscape.org/.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)