Keywords
solubility, molecular crystals, free energy calculations, chemical potentials, solvation
This article is included in the Cheminformatics gateway.
This article is included in the Mathematical, Physical, and Computational Sciences collection.
solubility, molecular crystals, free energy calculations, chemical potentials, solvation
Solubility is a critical property for pharmaceutical drug discovery; problems with solubility can frustrate drug discovery efforts and prevent treatments from working. The bioavailability of a drug depends on the solubility difference between different crystal structures (polymorphs), dose, drug permeability and formulation1, so solubility plays a key role. Solubility problems can be unexpected and can pose crucial obstacles that even threaten the administration of care. For example, a well-documented case occurred in the late 1990s, when ritonavir, an HIV-protease inhibitor marketed as Norvir, failed dissolution requirements2. Since ritonavir is not bioavailable in its solid form, it was administrated in capsules containing solutions designed not to be saturated with respect to the originally known molecular crystal (form I)2. The newly identified polymorph, form II, was unusually stable and unusually hard to crystallize; the preparation protocol of Norvir was inadequate to make capsules from the new polymorph, which severely threatened the supply of the drug in the market and endangered the lives of many HIV-poaitive patients2. Considerable effort has already been devoted to the methods to predict crystal polymorphs3–9, but much less attention has been given to methods to predict solubilities, with or without likely polymorphs as input.
Due to the importance of aqueous solubilities in different industrial processes and environmental applications, a scientific challenge, consisting of the prediction of 32 solubilities given a database of 100 reliable measurements10,11, was created with the goal of comparing the outcomes of different solubility prediction techniques. Participants employed methods such as artificial neural networks12, quantitative structure-property relationship (QSPR)13, and deep learning14 to predict the aqueous solubilities of drug-like molecules. All of the employed methods were empirical and trained on existing measurements. The limitation of these methods, however, is the dependence of a training set of data that limits their applicability to compounds similar to those in the training of the set and impairs its transferability.
Some newer methods attempt to predict solubilities based on a physical description of the interactions in solution and in the solid state, yielding results that are in principle rigorous given an accurate energy model and an adequate method. In these approaches, molecular systems are described using force fields, i.e, potential energy functions that contain parameters describing bonds, atoms, electrostatic and non-electrostatic interactions. Molecular dynamics or Monte Carlo simulations are commonly used to sample different configurations of the system described by an energy model called a force field. The simulations then allow the estimation of physical properties, such as internal energy, free energy, and enthalpy, under different conditions. The quality of the results of such methods depends on how well the force field describes the system under study and how good the sampling method is. Thus, some researchers have recently estimated aqueous solubilities using simulations of thermodynamic cycles encompassing the crystal, the ideal gas, and an infinitely dilute solution of a given molecule15,16. When the structure of the solid is unknown, some studies have substituted simulations of solid melts in place of a structure of the solid17–20.
While these physical methods for predicting solubilities have received some attention in the literature, most are still in their infancy, with only a handful of studies applying them, and it is not yet clear how broadly applicable they will be17–20, and others have only been suggested or demonstrated in proof-of-principle tests16,21–23. Our view is that the time is ripe for physical methods to predict solubility, especially given the routine nature of solvation free energy calculations at present24–29, which comprise essentially half of the solubility problem (see the Theory section). Polymorph and crystal structure prediction successes also mean that we may often have a suitable crystal structure of the compound as an input3–5,8,9,30–35, so what remains is to predict the solubility given a crystal structure and simulations of the relevant phases.
Here, we focus on adapting and testing an existing approach for solubility prediction in the hope that it will prove to be a generally applicable method for solubility prediction that can be applied routinely. This method uses all-atom molecular dynamics simulations to estimate absolute chemical potentials and predict aqueous solubilities of molecular solids, given the crystal structure (or an estimate thereof) as input.
Solubility is defined as the maximum concentration of solute that can be dissolved in a selected bulk solvent. Chemical potentials (µ) of the solid-state solute and the solution are by definition equal at the solubility point, when the solution is in equilibrium with the solid.
Solid particles precipitate in concentrations higher than the solubility point because the solid phase becomes more stable in these conditions. In principle, we can predict at which concentration a molecule precipitates in solution if we calculate the chemical potentials of the components:
where µi is the chemical potential of component i; A is the Helmholtz free energy; G is the Gibbs free energy; Nj, j≠i is the number of molecules of each component in the mixture; V is the volume of the system; T its temperature; and P its pressure. Calculations from systems under a constant V and T yield A; G is obtained from simulations under constant P and T conditions. In order to estimate the chemical potential of one component in solution and in its molecular solid, however, we need to know the absolute free energy of the system in these states. We calculated absolute free energies using alchemical free energy calculations.
The absolute free energy of a system can be determined if we know its partition function (Q), a function that connects microscopic properties of the system with macroscopic thermodynamic quantities. Unfortunately, it is very hard to calculate the absolute free energy of real systems because we don’t know their partition functions. Free energy calculations allow us to bypass this problem, but require at least two states: a reference state whose free energy can be analytically or numerically found, and a final state of interest36,37. We chose to calculate the free energy difference using alchemical free energy calculations, a method in which we simulate a series of non-physical intermediates between the end states38.
Each intermediate state in the alchemical path is described by a Hamiltonian ℋ (q, p; λ), i.e, the energy of the state as a function of atomic positions (q), momenta (p) and a coupling parameter (λ):
where ℋinitial and ℋfinal respectively are the Hamiltonians of the initial and the final state; and f (λ) and g(λ) are functions used to mix the Hamiltonians, and are usually set such that ℋ = ℋinitial at λ = 0 and ℋ = ℋfinal at λ = 1.
A variety of different estimators can be used to analyze alchemical free energy calculations, and have different strengths and weaknesses, as well as different data requirements. Here, we employ several different estimators we introduce briefly in the following.
One way to calculate the free energy difference (∆A) between the end states is Thermodynamic Integration (TI)39:
in which a set of discrete λ values correspond to states along the alchemical path. 〈〉 means that we are have to calculate the ensemble average of the derivative between the brackets. TI performs as well as more efficient methods if the integrand is smooth, but breaks down if this condition is not satisfied40–42.
An alternate free energy estimation method computes ∆A directly via:
where the ensemble average is calculated over the configurations of the initial state, and β is the reciprocal of kBT, the product between the Boltzmann constant and the absolute temperature. We call this approach exponential averaging43 (EXP).
Most free energy calculations involve many intermediates associated with the coupling parameter (λ), allowing simulation of intermediate states in between the two end states of interest. The free energy change between the end points of a path defined by N intermediates is:
where ∆An→n+1 is the free energy difference between (n+1)-th and the n-th intermediate states. Equation 5 can be used to calculate the free energy difference between each adjacent pair of states and yields the exact result at the limit of very large samples, but it is inefficient for most applications38.
The Bennett acceptance ratio44 (BAR) provides an estimator that is superior for most purposes. It calculates the free energy difference between the n-th and the (n + 1)-th states from the following relationship:
where Nn and Nn+1 are the number of statistically independent samples in states n and n + 1, respectively, and ∆ℋn→n+1 = −∆ℋn+1→n are the Hamiltonian differences between n and n + 1. BAR is more efficient than EXP45,46 and minimizes the free energy uncertainty44. Multistate Bennett acceptance ratio41 (MBAR) is an extension of BAR that takes in consideration the degree of configuration space overlap between a given state and all other states in the transformation, whereas BAR only uses the information of neighboring states. MBAR and BAR perform similarly when the spacing between the intermediate states is moderate, but MBAR is the most well-performing free energy estimator42.
In this work, we seek to predict the solubilities of molecular solids. Part of this problem requires predicting the free energy or chemical potential of the solid. One way this has been attempted in the past is via the Einstein crystal method (ECM), which calculates the absolute free energy of a solid using an Einstein crystal as a reference state. In this method, the crystal lattice is made of atoms restrained to their positions by a harmonic potential; additionally, the center of mass of the system is held fixed47.
In the ECM, and in this work, the absolute free energy of the molecular solid is found by designing a path where force field terms are progressively turned on, and the harmonic potential position restraints are turned off. The fixed center of mass is important to avoid a quasi-divergence issue when calculating the free energy term of releasing the system from the harmonic position restraints, but the contribution of the fixed center of mass needs to be included in the cycle to obtain the correct absolute free energy for the system (Figure 1(a))47–49.
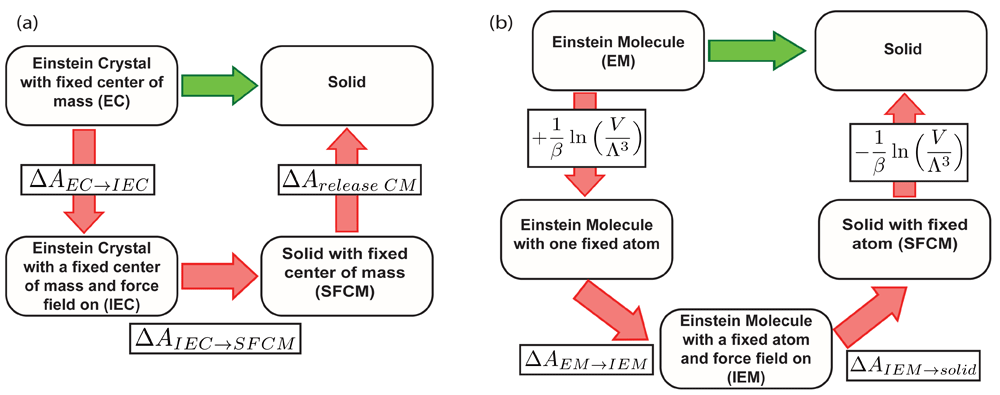
(a) Thermodynamic cycle representing the Einstein Crystal Method. (b) Thermodynamic cycle representing the Einstein molecule method (EMM). Note that the EMM requires only two free energy calculations despite being a bigger thermodynamic cycle. The canceling terms in (b) correspond to the free energies of fixing and releasing one atom in the crystal lattice48.
In ECM, the free energy is calculated by:
where is the free energy of the Einstein crystal (EC) with a fixed center of mass (FCM); ∆AEC→IEC is the free energy difference between the Einstein crystal (EC) and the interacting Einstein crystal (IEC), i.e., the free energy difference in a transformation where the force field is progressively turned on throughout the calculation path. ∆AIEC→SFCM is the free energy difference between the IEC and the solid with a fixed center of mass (SFCM), i.e, turning off the harmonic restraints; and ∆Arelease CM is the free energy of release of the center of mass (CM).
ECM can be difficult to implement because of the need for a fixed center of mass, so our work here is based on an alternative approach that is easier to implement. When particles move in ECM, the lattice needs to be moved because the center of mass is fixed47,48,50. Our method of choice, the Einstein Molecule Method (EMM, see Figure 1(b)), fixes a single atom in the lattice instead of the center of mass and is more easily implemented than ECM because of the relative difficulty of introducing center of mass restraints into existing simulation packages22,48,50–52. EMM has been used to predict phase diagrams of TIP4P and SPC/E water models48, free energies of ice polymorphs, solid methanol and toy systems49,52, and the solubilities of potassium and sodium chlorides22,51.
In EMM, the free energy of a solid is:
Asolid = AEM+∆AEM→IEM+∆AIEM→solid (9)
where AEM is the free energy of the ideal Einstein molecule; ∆Aid→IEM is the free energy difference between the ideal Einstein molecule and the interacting Einstein molecule (i.e, turning on the force field); and ∆AIEM→solid is the free energy difference between the interacting Einstein molecule and the solid (i.e, turning off the harmonic restraints). The advantage of EMM over ECM is the absence of the need to calculate a free energy term associated with releasing the fixed reference point48.
Here, as per Equation 9, we compute the free energy of the solid by combining the absolute free energy of the ideal Einstein molecule with two terms that we calculate via alchemical free energy calculations—∆AEM→IEM and ∆AIEM→solid; these involve alchemically changing the interactions in the system. Numerical integration of Equation 10 allows the calculation of the ideal term, AEM52:
where AEM and QEM are the free energy of the Einstein molecule and its partition function; UEM,1(Ω1) is the potential energy of the fixed particle 1; UEM,2(r2Ω2) is the potential energy of a non-fixed particle at a distance r2 of particle 1; Ω1 and Ω2 are all the possible orientations the molecules can have in the lattice; Λ, V, N, and β respectively are the de Broglie wavelength, the system’s volume, its number of particles, and the reciprocal of kBT, the product of the Boltzmann constant and the absolute temperature.
Another critical component of computing the solubility of a compound is estimating the chemical potential of a solute in solution, since the solubility point is the concentration at which the chemical potentials of compound in the two phases are equal.
The chemical potential of a component i in solution, µi, has an ideal and and excess component:
where qi is the internal partition function of a single molecule of the solute, U(Ni) is the potential energy of the system with Ni particles, Λ is the de Broglie thermal wavelength, and V is the system’s volume53. 〈〉initial means that the term was obtained from an ensemble average over the configurations of the initial state (see Equation 5). The first two terms of the equation above correspond to the ideal component of µi; the last one, , corresponds to the excess component of µi, and is associated with all non-ideal interactions of the extra component i with the solution (i.e. physical interactions that differ from those given by the ideal gas law). We obtained excess chemical potentials from solvation free energy calculations; the solute molecule is inserted in the solution by progressively turning on its interactions with the surrounding environment24,28,54.
The challenge associated with the calculation of µi is the calculation of the standard chemical potential of i, , the first term of Equation 11. qi, the internal partition function, includes the rotation, vibrational, electronic and nuclear partition functions of a single molecule53 and is unknown. Here, we found a way of calculating without the knowledge of qi by alchemically transforming a single solute molecule into a single Einstein molecule, whose absolute free energy we know how to calculate48–50.
We are aware of three main approaches to compute the solubility of solids in solution using physical approaches: ECM-based methods21,23, EMM-based methods22,51,55, and the approach of Michael Schnieders and collaborators which computes sublimation and solvation free energies and uses these in an alternate thermodynamic cycle to obtain solubility estimates15,56.
Many of the applications of these approaches have been to the solubility of ionic solids, with both ECM-21 and EMM-based approaches22,51,55 having some success. However, molecular solids introduce substantial additional complexities for both of these approaches.
The ECM has seen an initial test on solubility estimation. Li et al.23 used the ECM to estimate the solubility of napthalene, but made several approximations such as assuming that the internal partition function component of the solute cancels between environments (perhaps justified given napthalene’s low solubility).
We are not aware of any work applying the EMM to solubility estimation of molecular solids; to our knowledge our work is the first to make such an attempt, though EMM has been used before to estimate the free energy of simple molecular solids49,52 but not the solubility. This explains our need to find our own approach to estimate for a single solute molecule.
The Schneiders approach is an orthogonal one that we do not examine here.
Here, we chose three systems to study: An argon crystal for some small initial tests, α-methanol to help establish our protocol, and acetylsalicylic acid (ASA) as our main object of study. ASA is a known anti-inflammatory whose most stable polymorph, form I57, has an aqueous solubility of approximately 0.038% mole fraction at 298 K58. We also used α-methanol at 150 K and a toy facecentered cubic (fcc) argon crystal59 to help us find an optimal protocol to calculate the absolute free energy of a molecular solid. α-methanol was chosen because it had been used before in a study that applied the EMM to calculate the absolute free energy of the solid52.
All simulations were run in GROMACS 4.6.760–63. With one exception, all simulations used the General Amber force field (GAFF) version 1.7 with AM1-BCC charges64,65; the exception was α-methanol, because we ran these simulations using the input files – coordinates and force field parameters – provided by Aragonès et al., who used an united atom version of the OPLS force field52.
We simulated all solids and liquids using 5 ns Langevin dynamics simulations. ASA, α-methanol, and argon were simulated respectively at 298.15 K, 150.0 K, and 4.0 K. Our simulations had the same length as the simulations run by Aragonès et al. All solid state simulations were run in NVT conditions. Liquid state simulations were run in NPT conditions; pressure was kept constant at 101.335 kPa using the Parrinello-Rahman barostat66. We used the TIP3P water model67 for all our liquid state simulations. More simulation details and example input files with full details can be found in the Supporting Information.
The absolute free energies of the solids were calculated from trajectories of simulation boxes with 64 ASA molecules, 100 OPLS methanol molecules, and 864 argon atoms with periodic boundary conditions. ASA’s unit cell was obtained from Mercury CSD 3.868 and the fcc argon crystal was obtained from the literature59. Simulation box sizes were chosen to be approximately between 2 nm and 3 nm to ensure that box sizes were large enough that atoms and their periodic copies were not within cutoff distance of one another. α-methanol’s crystal was obtained from the Supporting information of Aragonès et al.52 We used Amber14’s ambertools69–72 and ParmEd73 to generate the ASA’s and argon’s solid state input files. All atoms but one were subjected to harmonic constraints in the x, y, and z coordinates. A single atom was kept fixed in space to act as the reference point for the calculations, as explained in the Introduction.
Monte Carlo integration yielded AEM, the free energy of the Einstein molecule, as it was previously done for α-methanol in the literature52. ∆Aid→IEM and ∆AIEM→solid were estimated using TI39 and the multistate Bennett acceptance ratio (MBAR)74. We used force constants of 4000 kBT/Å2 to restrain atoms to their lattice positions in ASA and argon simulations because it allowed us to use a reasonable time step of 1.0 fs in all simulations. α-methanol simulations used the same force constant that had been previously used by Aragonès et al.52.
We used alchemical free energy calculations to obtain the difference in free energy between the reference Einstein molecule and the solid. This step was divided in two parts: (a) the force field parameters are alchemically turned on, and (b) the harmonic constraints are turned off.
Here, we deviate from earlier work which calculated the absolute free energy of a solid using EMM by introducing additional intermediate states to improve accuracy, along with using a superior free energy estimator.
For the calculation of ∆Aid→IEM, we found it was crucial to introduce intermediate states; we also switched to using the MBAR estimator. The original EMM calculation of the absolute free energy of a solid22,48–52 estimated ∆Aid→IEM using exponential averaging (EXP) with just two states: the Einstein molecule (EM) and the interacting Einstein molecule (IEM)21,22,48–52,55. As EXP is known to have convergence issues and biases38,40,41,45, we switched to the superior MBAR free energy estimator74. Additionally, when we did so, we found that overlap of states (as measured by the overlap matrix75) was insufficient so we created a series of intermediate states connecting both ends of the transformation.
For ∆AIEM→solid., the original work used TI39. Here, we replaced TI with MBAR as our analysis method of choice. Generally, the literature shows that TI performs as well as more efficient methods like BAR and MBAR when the integrand is smooth38,40,41, but it is sensitive to the choice and number of intermediate states76. MBAR is the most consistently well-performing free energy estimator42 and exploits the overlap between states more thoroughly than its predecessor, the Bennett Acceptance Ratio (BAR) estimator74. Here, we chose to compare performance of MBAR and TI for calculation of ∆AIEM→solid for ASA and α-methanol; we also applied EXP as a comparison in the latter case only.
The chemical potential of a pure solid is its molar free energy:
where N is the number of molecules in the solid, and A its Helmholtz free energy.
The chemical potential of a substance i in water is defined as the derivative of the free energy of the system with respect to the composition:
where G is the Gibbs free energy, and Ni is the number of molecules of i in solution; P, T, and NH2O are the pressure, absolute temperature, and number of water molecules in solution, and are kept constant in the calculation.
One important aspect to discuss is the reason why we chose to calculate the Helmholtz free energy for the solid and Gibbs free energies for each solution. Solid state simulations with position restraints required running under constant temperature and constant volume conditions due to software limitations, therefore we were able to calculate A for the solids. At constant pressure, both kinds of free energy are related by:
∆G = ∆A+ P∆V (14)
Since solids are much less susceptible to volume changes than liquids, it is reasonable to consider that P∆V is negligible and ∆G ≈ ∆A. For instance, the difference in volume between the experimental ASA crystal structure and the simulation box after a constant pressure equilibration stage is 0.14 nm3. The P∆V term – i.e., the free energy difference discounting possible structure relaxation effects – would be much smaller than the simulation error.
As we explain in more detail in the Results section, successful absolute free energy calculations for molecular solids require a pathway involving a large number of alchemical intermediate states. The calculation of the absolute free energies of α-methanol at 150 K and ASA required 600 states. Since GROMACS reads each λ until its fourth decimal place and the states need to be spaced more closely together as as the harmonic restraints are turned off (see Supporting Information), we decided to split each free energy calculation into sets of 100 states.
Liquid state simulation boxes were generated using the SolvationToolkit77, a Python package that uses packmol78, OpenMolTools (v0.6.7)79 and OpenEye Python Toolkits80–82. Excess chemical potentials were obtained with the same solvation free energy protocol used in previous studies28: Starting from a fully interacting system, we progressively decouple the interactions of a single solute molecule with the remaining of the system, which allows us to calculate the free energy difference between a solute molecule in vacuum and in solution (i.e., the solvation free energy).
We also used alchemical free energy calculations using a single Einstein molecule as a reference state to estimate the standard chemical potential of a substance, :
where and respectively are the chemical potential associated with turning off the force field and chemical potential of restraining the atoms of the molecule to their lattice positions. is calculated using the Monte Carlo integration procedure that we used to calculate AEM to a single molecule.
The first step to predict aqueous solubilities with the aid of absolute free energy calculations was the assessment of the methodologies we chose to use. Since our method is the same one used by Aragonès et al.52 and we wanted to be sure that we could reproduce previous results, we ran simulations for α-methanol at 150 K and estimated the free energies of solids using MBAR. Turning off the harmonic restraints was the challenging step. Our MBAR calculation of ∆AIEM→solid for α-methanol using 18 intermediate states yielded −18(3) kBT, while our TI result was −18.421(5) kBT and the literature result was −17.33(3) kBT using 17 states52. The MBAR error was unusually high (3 kBT), which is usually a signal of overlap problems or other serious concerns.
MBAR is a free energy estimation method that minimizes the free energy variance and considers the overlap between a given state and all the others in the transformation path41, which means that high uncertainties (±3kBT) suggest the presence of problems in the transformation’s path. TI’s uncertainty estimates are much lower, but we believe that this is an artifact. Error analysis for TI simply does not work the same way and does not give insight into whether exploration of phase space is adequate, unlike MBAR. Specifically, uncertainty estimates from TI usually factor in only the uncertainty in the integrand at each sampled lambda value and could potentially also factor in the smoothness of the integrand (i.e. numerical integration error) but do nothing to factor in whether the integrand will in fact vary smoothly in between lambda points; usually no data is available on this. BAR and MBAR, in contrast, factor in information about how well the intermediate states overlap in phase space and reflect high uncertainties when phase space overlap is poor. In our experience, TI would usually suffer from similar problems if additional intermediate states were added, but uncertainties in TI typically do not reflect this, as is the case here. Thus, the high uncertainty of the MBAR value indicates a sampling/convergence problem which warrants further exploration.
To explore the high uncertainty of our MBAR free energy estimates, we examined the degree of overlap the intermediate states had with each other. Phase space overlap analysis83–85 quantifies the probability that any given configuration of an intermediate state can be found in other states. A good rule of thumb for designing a set of free energy calculations spanning between two states is to ensure that the states along the path have significant overlap with their neighbors as shown in Figure 2. More overlap improves the quality of the MBAR free energy estimation: Figure 2b represents a set of restraining simulations where the free energy uncertainty can potentially be accurately estimated using BAR and MBAR; Figure 2a shows a case where it cannot. In our case we find that the α-methanol simulation using 18 intermediate states does not have adequate overlap (Figure 3)– specifically, the states 4 ≤ λi ≤ 17 do not have overlapping configurations with other states, which explains the 3 kBT uncertainty in our MBAR estimate.
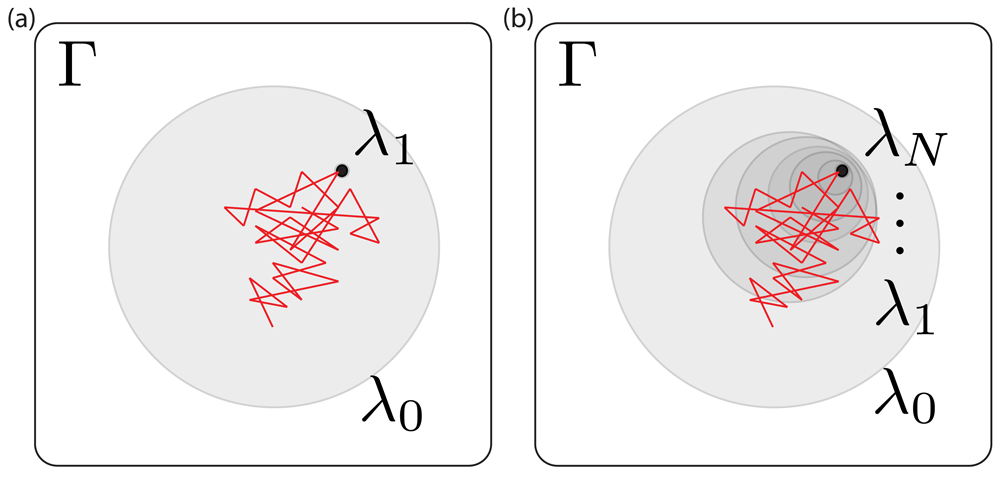
Γ represents the phase space that contains all the configurations for all the states in the path. λ0 and λ1 (left) or λN (right) represent the end states along the path, each shaded region represents a state in phase space and the red lines represent the configurations visited by the simulation run in the λ0 state. The restrained state is a subset of the unrestrained one. (a) and (b) represent simulations with different numbers of intermediate states along the path between a fully restrained state (λ1 (a) or λN (b)) and an unrestrained state (λ0). In (a), the simulation (red) only visits very few configurations consistent with the restrained state – i.e, there is poor phase space overlap – indicating a need for more intermediate states, otherwise any free energy estimates will be subject to very high uncertainties; in (b) there is still almost no overlap between the simulation and states consistent with λN, but there is overlap with the next shaded region, λ1, indicating the potential for overlap and accurate free energy estimates. Thus simulations run in each shaded region are more likely to have a bigger phase space overlap with λN than simulations run in λ0.
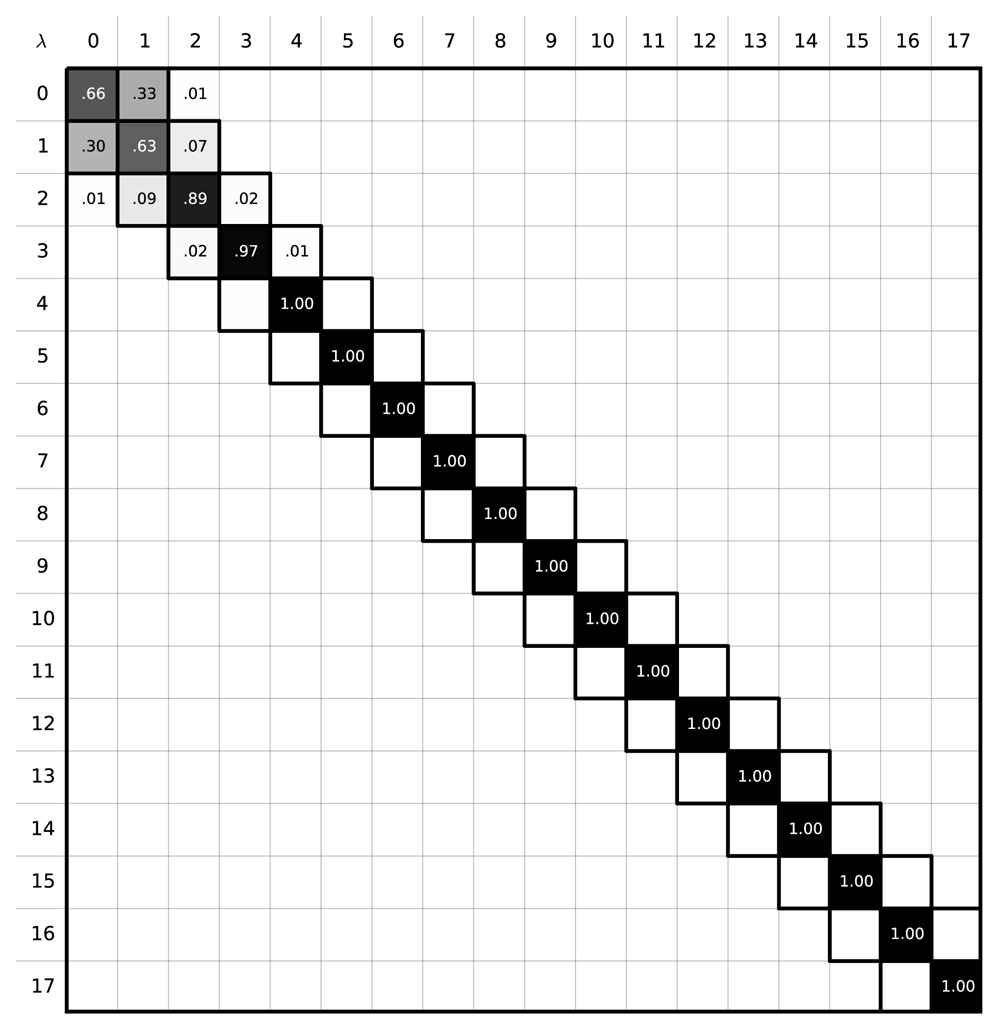
The sum of all the elements in a row should yield 1.0, a probability of 100%. A good free energy estimate is obtained when the states along the alchemical path contain configurations that can be found in other intermediate states. In these situations, the phase space overlap is non-zero, which results in non-zero off-diagonal elements. Here, however, the phase space overlap plot shows that there is no overlap between the states λi, 4 ≤ i ≤ 17 indicating poor free energy estimates will result.
Since prior work had appeared to do this estimation successfully52, we were uncertain why we were encountering such overlap problems, so we studied an even simpler system. We calculated ∆AIEM→solid of fcc argon at 4 K with 18 states as in our α-methanol free energy estimation. MBAR yielded an error estimate of infinity, whereas TI estimated ∆AIEM→solid to be −1666.5(8) kBT, which, as we show below, is incorrect. This path resulted phase space overlap diagram without overlap between the states after state number 2 (Figure 4). Apparently as the harmonic potential that holds atoms in their lattice positions tends to zero, atoms become rather mobile, dramatically decreasing phase space overlap and leading to poor free energy estimates.
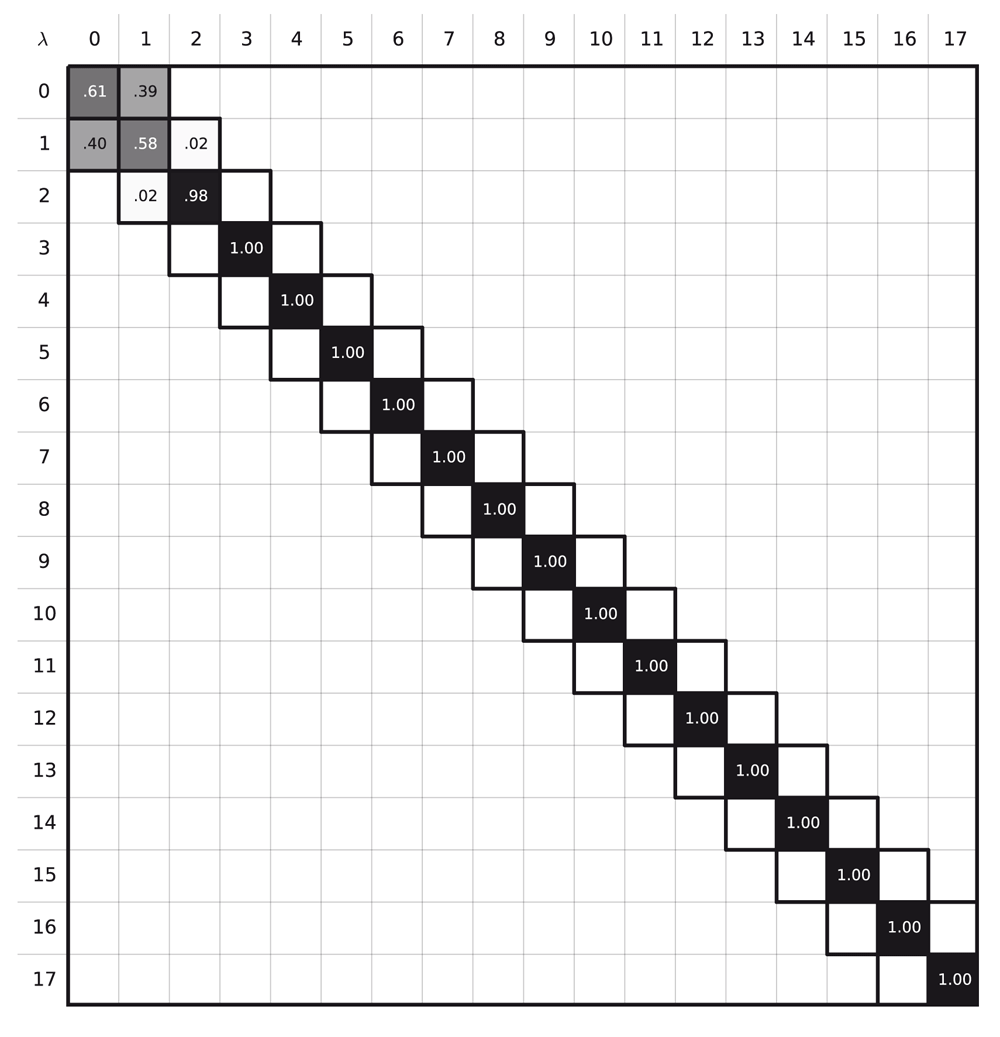
A good free energy estimate is obtained when the states along the alchemical path contain configurations that can be found in other intermediate states. Here, however, the phase space overlap diagram shows that there is no overlap between the states λi, 3 ≤ i ≤ 17, which explains the poor quality of the free energy result.
To improve phase space overlap, we introduced more intermediate states along the path for removing the restraints (see Figure 2). We chose to break down the simulation in smaller parts, adding a significant amount of states near the point where the harmonic restraints are approximately zero. The MBAR estimate of ∆AIEM→solid for fcc argon is −1016.0(2) kBT using 300 states. TI’s corresponding value was −1017(1) kBT, differing by far from the (incorrect) value of −1666.5(8) kBT obtained above with fewer states. Phase space overlap diagrams showed significant improvement in the configuration overlap between the states (Supporting Information). Thus, increasing the number of states was an effective strategy, and we used it in all subsequent calculations.
Even though our α-methanol results were similar to results published previously by other authors52, we need to emphasize that reliable free energies resulted from simulations with a large number of intermediate states, as can be seen in Table 1. Despite its conceptual simplicity, calculating the components of the absolute free energy of a solid to a point where there is significant phase space overlap between the intermediate states is computationally demanding. A 900-atom OPLS α-methanol system required 40 states to calculate ∆Aid→IEM, and 600 states for ∆AIEM→solid.
| Literature52 | Our replica | |
|---|---|---|
| AEM | 29.05 | 29.24 |
| ∆Aid→IEM | −41.27(1) | −38.04(7) (EXP) −41.306 56(4) (MBAR, 20 states) −41.275 719(7) (MBAR, 40 states) |
| ∆AIEM→solid | −17.33(3) | −18.421(5) (TI, 18 states) −18(3) (MBAR, 18 states) −17.1712(6) (TI 600 states) −17.1692(4) (MBAR, 600 states) |
We chose these intermediate states in advance, and these ultimately led to free energy errors smaller than 0.1 kBT; the estimated TI and MBAR values differed by no more than 0.3 kBT. Our results for ASA using an optimal number of states can be seen in Table 2. The MBAR chemical potential of ASA at 298.15 K equals to −220.67(3) kBT.
| Acetylsalicylic Acid | |
|---|---|
| AEM | 48.047 |
| ∆Aid→IEM | −167.316(1) (TI, 118 states) −167.07(3) (MBAR, 118 states) |
| ∆AIEM→solid | −101.656(2) (TI, 600 states) −101.644(2) (MBAR, 600 states) |
The computational cost of calculating AASA was high; Each state required a separate simulation (of a 1344-atom ASA system), with 718 states in total. Simulations typically required 11 hours on a single CPU, so the calculation of a single absolute free energy of a molecular solid required approximately 7898 CPU-hours.
Equation 11 states that the absolute chemical potential of a solution is determined by three quantities: , the standard chemical potential; , the excess chemical potential of the component at a concentration of χ; and a volume-dependent ideal gas component of ln (NASA/〈V〉solution). Calculation of only required information regarding the internal structure of the molecule53, thus we estimated by alchemically transforming a single solute molecule into a single Einstein molecule (Table 3), whose absolute free energy we know how to calculate. We used the same number of states that we chose for the solid state simulations and we found that is equal to –150.7(2) kBT, as discussed in the last subsection of the Methods section.
| Acetylsalicylic Acid | |
|---|---|
| 9.3 | |
| 65.7409(9) (MBAR 118 states) | |
| 94.3(2) (MBAR 600 states) |
Concentrations, volumes and excess chemical potentials can be seen in Table 4. We obtained the excess chemical potentials from solvation free energy calculations24,28,54. Volumes were obtained from the state in the alchemical path where the solute was fully coupled to the rest of the system.
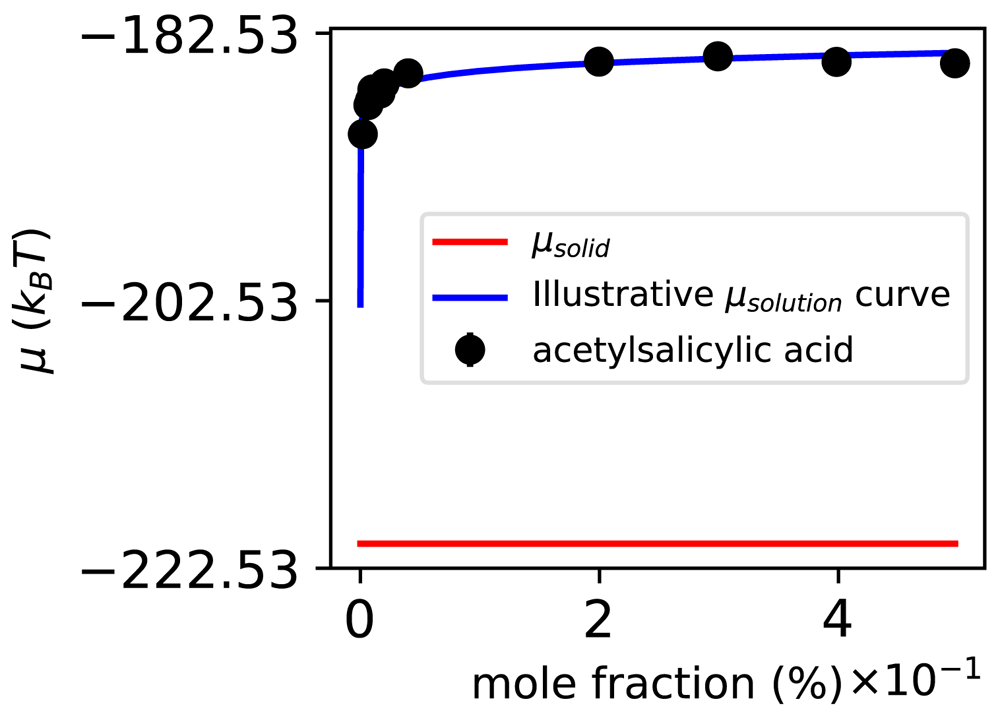
The experimental aqueous solubility of ASA is approximately 0.038% in water at 298 K58, but our model predicts that ASA is effectively insoluble in water (Figure 5). While all-atom simulations can yield solubility estimates given adequate simulation time and a correct method, the computed solubility will be that dictated by the underlying energy model or force field, and will not necessarily match experiment. Here, we use GAFF, a general-purpose force field with known limitations28,71,86,87; apparently, here, the right answer for the force field is not correct. Perhaps this is because of limitations in describing the solid state, as the force field is parameterized for liquid state simulations. Indeed, classical fixed charge force fields have shown severe limitations for polymorph prediction for these reasons5,31,33–35. Also, point partial atomic charges regularly used in molecular dynamics do not describe electrostatic interactions in a solid particularly well88. In the case of the ASA crystal, it is possible that its hydrogen bonds and π-stacking interactions add layers of complexity that are not properly described by GAFF.
Despite its theoretical rigor, solubility prediction from absolute free energy calculations is a difficult task: it is computationally expensive and, at least in the present approach, requires many different steps and a great deal of care. Here, we attempted to develop and test a general approach to compute the solubility of molecular solids by adapting the EMM to tackle this problem, as discussed above.
To tune our methodology, we initially decided to reproduce the absolute free energy of solid α-methanol, one of methanol’s polymorphs, at 150 K using EMM before doing the same calculations for our compound of choice, ASA. We verified that the free energy differences between the Einstein molecule and the interactive Einstein molecule (∆AEM→IEM) and between the latter state and the solid (∆AIEM→solid) were more reliably estimated with the MBAR. The absolute free energy of the crystal (as computed for united-atom OPLS α-methanol) agreed with results found in the literature, which suggested that we were on the right path. We did, however, require a very large number of intermediate alchemical states to obtain accurate free energy estimates, making these simulations fairly computationally demanding
We then chose to calculate the solubility of ASA, owing to its pharmacological importance and its relative complexity compared to previous molecular solids, whose absolute free energies have been computed via EMM previously52. As for α-methanol, this calculation required a large number of intermediate alchemical states and considerable computational cost – approximately 8000 CPU hours for a single absolute free energy calculation for the molecular solid, even with the crystal structure as input. Perhaps the number of intermediate states could be further optimized, but clearly a large number of intermediate simulations was required and thus considerable computational cost. Despite all of this, we still could not reproduce the experimental aqueous solubility of ASA; experimentally it is modestly soluble, whereas our work would suggest it is essentially completely insoluble in water, likely due to force field limitations.
The solubility of naphthalene was recently estimated using a similar methodology, the Extended Einstein Crystal Method23, but with additional approximations. Specifically, since naphthalene molecules interact very weakly with each other in the crystal lattice and with water molecules in solution, the differences between the internal partition function of a naphthalene molecule in the solid and in the solution were assumed to be negligible. This allowed the authors to drop some complexities in treatment of the solution-phase part of the calculation. However, that approach is only suitable for compounds that are only very weakly interacting in solution and in the crystal. ASA, in contrast, is a molecule that interacts strongly with other ASA molecules in its crystal lattice and with water molecules in solution via hydrogen bonds. For instance, an important crystalline feature that is not necessarily present in solution is the dimer structure, with two ASA molecules bound together via hydrogen bonds between the carboxylic acid groups. Differences between the internal partition functions of the molecule in the solid and in solution would probably not be negligible in this scenario, thus a more general approach is needed for treatment of such cases. Our work here provides one attempt in that direction.
Overall, the present approach seems to have significant limitations – most notably that the computational expense is considerable, and the resulting estimated solubility is quite inaccurate. Perhaps both of these may be surmountable; GPU-based free energy calculations can be dramatically faster, potentially reducing an 8000 CPU-hour calculation to 80 GPU hours, which would amount to overnight on 8 GPUs, and perhaps this could be optimized via changes to simulation time and number of intermediate states. And with better force fields, perhaps accuracy could be improved; the AMOEBA-based approach of Schnieders shows considerable promise15. Alternatively, other approaches may be of interest. Solubility has been predicted by simulations using pseudocritical paths (i.e., paths were molecular crystals are transformed in tractable Einstein crystal-like states between the ending states of the transformation89–92) and a single experimental reference point91), and with the aid of a thermodynamic cycle formed by the molecular crystal, the molecule in vacuum, and the solvated molecule15.
We believe the time has come for routine physical methods for estimation of solubility, even if improved force fields prove necessary before results have significant accuracy for application to biomolecular design problems.
All data underlying the results are available as part of the article and no additional source data are required.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)