Keywords
Inflammatory Bowel Disease, Depression, Microbiome, Machine Learning, Random Forest, Metagenomic, Metatranscriptomic.
This article is included in the Bioinformatics gateway.
Inflammatory Bowel Disease, Depression, Microbiome, Machine Learning, Random Forest, Metagenomic, Metatranscriptomic.
Main difference from previous version and the version 2 is that as per Reviewer 1’s comments, we have added a more thorough description of the dataset and a workflow illustrating the k-Fold Cross Validation approach for Random Forest (new Figure 1). As per the comments of Reviewer 2, we have rerun the species identification considering all IBD samples as a group and then classified by a depressed or not depressed state. We have furthermore added a small description of the set of species and expanded the introduction with some of the citations suggested. However, we cannot add covariation analysis as the metadata is quite incomplete and heterogeneous and likely to give misleading results - hence this was not performed.
See the authors' detailed response to the review by Yasir Suhail
Increased depression rates have been frequently reported on patients with inflammatory bowel disease (IBD) (Graff et al., 2009), which is a big concern from a clinical standpoint, since increased levels of stress and anxiety are major drivers of IBD relapse and severity (Mawdsley & Rampton, 2006). Both IBD and depression are heavily influenced by the gut microbiome structure, which controls anti-inflammatory processes and permeability in the gut, and communicates with the brain by a complex and close relationship with the Autonomous Nervous System that is known as the brain-gut axis (Foster & McVey Neufeld, 2013; Luna & Foster, 2015; Martin et al., 2018).
Altered microbiomes can have big impacts on the health and development of both the gut and brain, and alterations in the ecology of this microbiome, a process known as dysbiosis, have been separately linked to both depression and IBD (Kaur et al., 2011; Rogers et al., 2016). While IBD has become one of the main focus on microbiome research for its clinical relevance and complex relationship with metabolic, immune and neurological processes (Huttenhower et al., 2014), research on the effect of the microbiome in mental health are comparatively scarce, but have already shown promising results reducing anxiety and depression symptoms using probiotics (Bravo et al., 2011; Pinto-Sanchez et al., 2017). However, the relationship between different microbiome population structures and these conditions is still poorly understood.
The availability of the large amount of data derived from the recent explosion in metagenomics and metatranscriptomics provides unique opportunities for investigation. However, it is sometimes difficult to identify informative species. Recently, machine learning algorithms have been successfully applied because they allow the identification of patterns in situations where large, multi-dimensional and heterogeneous datasets are available.
Among the several machine learning approaches available, random forest is an algorithm used for classification and regression based on an ensemble that builds a population of decision tree classifiers, such that the result of a prediction from a given set of features is the most frequent result from the different trees of the “forest” (Breiman, 2001). This is an efficient and generalist algorithm that has already been applied in several metagenomic investigations in human diseases, such as IBS (Saulnier et al., 2011).
The aim of this work was to apply the random forest approach to identify the microbiome species that may be mostly involved in IBD and depression outcomes and that are responsible for the most relevant changes in the population structure between IBD, depression and patients comorbid for both conditions, and to provide insights on how the microbiome is involved in this comorbidity.
The datasets used for the analyses were retrieved from the Inflammatory Bowel Disease Multi-Omics Database (IBDMDB) (Schirmer et al., 2018), which is part of the Integrative Human Microbiome Project (NIH HMP Working Group et al., 2009). The IBDMDB database contains a wide array of omics data (e.g., 16S and shotgun metagenomic, metatranscriptomic, proteomic and host genomes) of 132 individuals classified by IBD diagnostic in ulcerative colitis, Crohn’s disease and controls. Participants provided bi-weekly stool samples at five hospitals in the United States. Metagenomic and metatranscriptomic data was processed as described in Schirmer et al., 2018 (Abubucker et al., 2012; Truong et al., 2015).
From this dataset, the 70 unique participants who answered an additional self-reported depression and anxiety questionnaire during registration (the answers to which are listed in the HMP2 metadata, column EC to EL) were selected. As the questionnaire model was not specified, only individuals with raw scores over 6 on this test was considered as showing “signs of depression”. To calculate the raw scores, a severity scale was generated, with the following scores: 0, never; 1, rarely; 2, sometimes; 3, often; 4, always. The scores were then summed to give a final total. In the case of individuals undergoing multiple tests, the lower score was used. We selected a low threshold in order to be able to identify putative dysbiotic individuals that were not experiencing severe depression symptoms. All the others were classified as “no sign of depression”. The combination between the test and the IBD diagnosis divided the dataset in six groups: Crohn’s disease with no detectable sign of depression (CD; n=15), Crohn’s disease with signs of depression (CDD; n=20), ulcerative colitis with no sign of depression (UC; n=4), ulcerative colitis with signs of depression (UCD, n=11), signs of depression but no inflammation (nonIBDD; n=7) and the control group: no inflammation/no depression (nonIBD; n=13). As the experimental design of the IBDMDB consisted of a longitudinal study, each subject contributed several times to this study, and all the samples used for this analysis were sequenced by shotgun sequencing as described in Schirmer et al. The resulting datasets for metagenomic and metatranscriptomic consist of 1084 and 566 samples, respectively. The final tables after pre-processing consist of 1486 columns, including Participant ID, data type, diagnostic, sex, mental score, and nested columns on the relative values of the different taxa.
For each of the six groups, abundance matrices of the metagenomic data, metatranscriptomic data, and the combination of metagenomics and metatranscriptomics were used for random forest classification. Each of the datasets was divided randomly into a training set (90% of the individuals) and a validation set (10% of the individuals). Random forest analysis were performed using the library Scikit-learn 0.19.1 (Pedregosa et al., 2011) on the training sets to identify the most important species involved in discriminating the samples without losing predicting power. A 1000-fold cross-validation for the combined dataset, and 500-fold for metagenomic and metatranscriptomic data (see Figure 1), considering one model for each iteration was performed and only the most important species in the construction of this model was retained. Only models with a precision classification >80% were considered, and among the considered models, only species that appeared more in more than one were selected. Afterwards, the validation sets were run with the selected species only to measure the possible loss of predictive capability and computed the area under the receiver operating characteristic (auROC) curve for the prediction of the validation set classes as a performance metric.
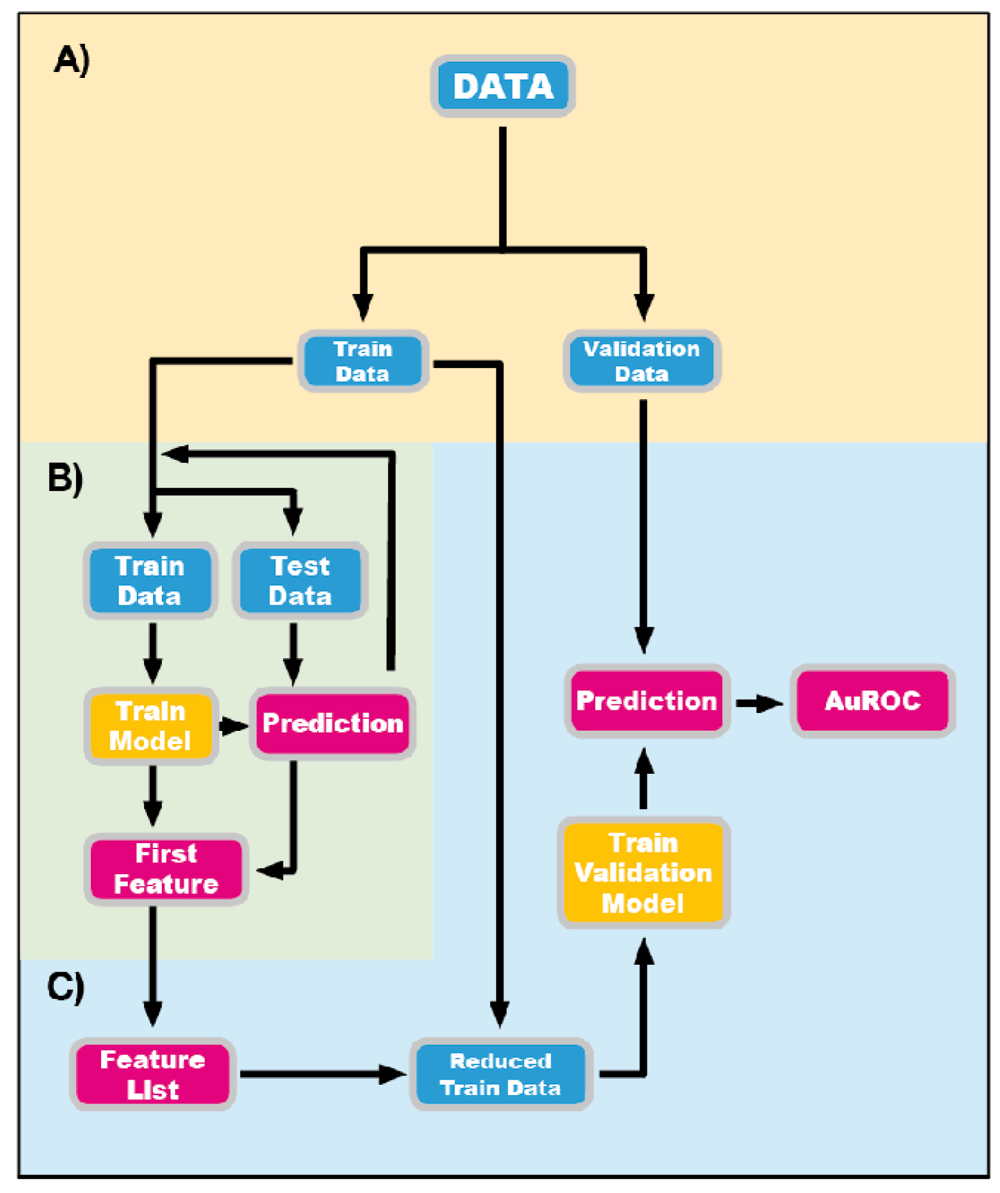
First the data gets split into train and validation sets (A). The train dataset will be iterated by the Cross Validation algorithm (B), while the validation set will be spared to test the model trained only with the reduced feature list (C).
In order to assess the significance of the differences between the abundances of the selected species, we performed a one-way ANOVA (Scipy 1.0.0, Jones et al., 2001) with a Tukey’s honest significant difference (HSD) post-hoc test. This test makes pair-wise comparisons between the different means to see which classes are different. For clarity, confidence intervals for Tukey’s HSD test can be found in Supplementary Materials (Supplementary Figure 1 and Supplementary Figure 2).
The functional activity of the selected species was retrieved from the HUMAnN metatranscriptomic analyses described above. Only the pathways in which the selected species are involved and those that were different between the groups from the ANOVA test were selected and the correlation between these species was calculated using Spearman’s correlation coefficient. A significance level of 0.05 was applied for all statistical tests.
The random forest cross-validation selection of the most informative species showed a combined list of 24 species, as can be seen in Figure 2. The validation models for DNA, RNA and the combined dataset shows micro-averaged auROC values of 0.96, 0.91 and 0.99, respectively (Supplementary Figure 3–Supplementary Figure 5). This small loss of information suggest a relevant role of the selected species in the interaction of both conditions, while the capability of the model to classify the validation data with with great accuracy shows that our model can generalize its results and it’s not overfitting.
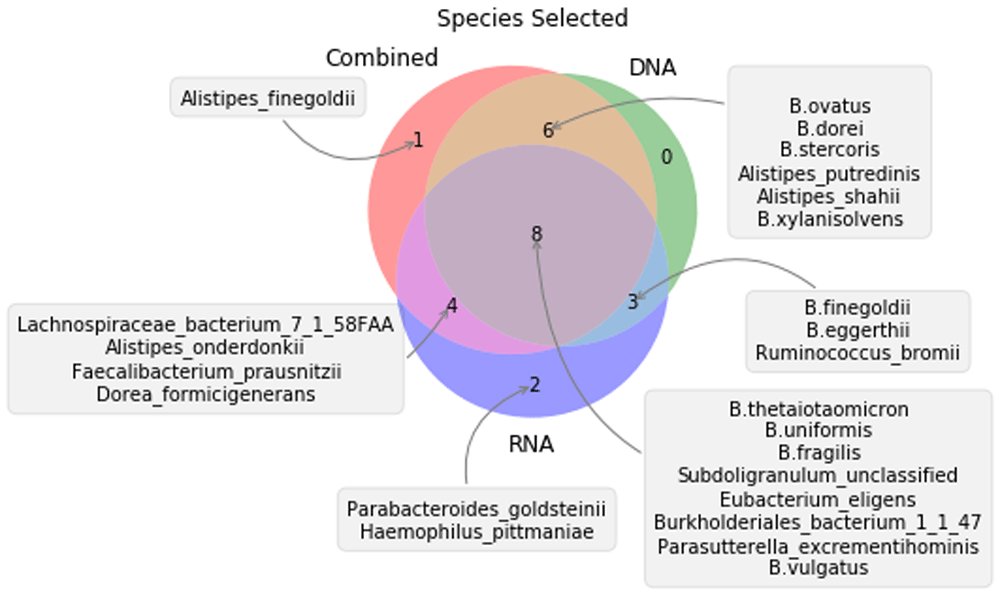
All species exhibited differences in at least one group in a one-way ANOVA (alpha=0.05, Supplementary Table 1), and no significant differences were found between DNA and RNA abundances for these species (Supplementary Table 2). This list of putative species pretends to be a trade-off between the all-relevant and minimal informative approaches. We chose this approach ir order to get as broad of a list as possible while avoiding artifacts related to the longitudinal nature of the dataset.
In order to assess the effect of the small sample size of group UC, the same procedure was made grouping all samples with IBD together. As expected, we see some difference in the species selected. However, the species that showed stronger differences in the previous classification were also the stronger ones, with most of the species overlapping. The interesting exception is Faecalibacterium prausnitzii that was absent.
The analyses showed an increase in the number of species from the genus Bacteroides in dysbiotic groups compared with the control (nonIBD) (Figure 3), as has been reported in other dysbiotic samples (Bloom et al., 2011), with the exception of Bacteroides dorei, which is more abundant in nonIBD than in any other group. Aside from Bacteroides dorei, nonIBD samples had a higher abundance of Alistipes shahii and Ruminococcus bromii, while a typical species associated with nonIBD, Faecalibacterium prausnitzii, was significantly decreased in nonIBDD and CD.

DNA (A) and RNA (B) taxonomic abundances for the selected species. Abundances were quantified by the relative abundances of their sequences, and for each level they should sum to 1 (including unclassified sequences).
Both of the Crohn’s disease-related groups (CD and CDD) showed higher abundances of Bacteroides ovatus and Bacteroides uniformis. However, CD samples exhibited higher abundances for several specific species, including Bacteroides xylanisolvens, Parasutterella excrementihominis and Bacteroides fragilis, compared with CDD, but decreased abundance of Faecalibacterium prausnitzii, which did not differ significantly in abundance between nonIBD and CDD groups.
Ulcerative colitis samples had the most distinctive microbiome profile. Several species, including Burkholderiales bacterium 1_1_47, Bacteroides eggerthii and Bacteroides finegoldii were characteristic of this group, and absent in the others, except for B. finegoldii, which was also present in a lower abundance in nonIBD samples. Only UCD samples exhibited an increased abundance of Bacteroides fragilis, Bacteroides vulgatus and Haemophilus pittmaniae, this last species being almost exclusive to the UCD group.
The nonIBDD was the group with the highest number of changes in microbiome diversity when compared with its non-depressed counterpart (Table 1). However, most of those changes followed a similar pattern in other dysbiotic groups.
Increases/decreases shown are statistically significant.
A notable change was observed in Faecalibacterium prausnitzii, which was present in almost the same abundances in nonIBD, UCD and CDD samples, and a high variability in UC while being significantly lower in CD and nonIBDD (Supplementary Table 3 and Supplementary Table 4). This is particularly interesting, since this species is considered to have anti-inflammatory activity. It seems counterintuitive to find a depleted population of one of the species most associated in the literature with a healthy microbiome compared to an IBD one in a group that doesn’t show any inflammatory process. However, Parabacteroides goldsteinii was increased in nonIBDD and was depleted in all IBD groups in comparison with control samples. The Parabacteroides genre have been associated previously with anti-inflammatory activity (Neff et al., 2016; Schirmer et al., 2016), so the increase in abundance of this bacteria may explain why the nonIBDD microbiome is not associated with inflammation in the gut.
Other than Parabacteroides goldsteinii, nonIBDD samples did not contain other characteristic groups, and, more notably, none of the selected species was specific for depressed or non-depressed phenotypes.
Regarding the functional activity of these species, seven pathways that were more abundant in dysbiotic groups than in nonIBD were identified (Supplementary Figure 1) and were correlated between each other and inversely correlated with most of the others (Supplementary Figure 2 and Supplementary Table 5). Those pathways are folate transformations II, N10-formyl-tetrahydrofolate biosynthesis, de novo L-ornithine biosynthesis, superpathway of pyridoxal 5’phosphate biosynthesis and salvage, phosphopantothenate biosynthesis I, preQ0 biosynthesis and queuosine biosynthesis. Folate (vitamin B9) and pyroxidal 5’-phosphate (vitamin B6) deficiencies have been linked both to depression (Coppen & Bolander-Gouaille, 2005; Hvas et al., 2004; Mitchell et al., 2014), as they are key for the synthesis of several neurotransmitters, and IBD (Pan et al., 2017; Yakut et al., 2010), although this association is not well understood and does not seem to be evidence of causation. Increased levels of L-ornithine derivatives have also been linked to depression (Zheng et al., 2010). However, even if nonIBDD have the highest activity for almost all of these pathways, CD and UC were also significantly increased, while functional activity in CDD was generally lower and non-significant in some pathways. Moreover, UCD did not differ from nonIBD in any of them.
This difference in functional activity again highlights the lack of a concrete pattern of gut microbiome abundance between depressed groups.
The random forest approach was able to successfully identify informative changes in abundance at the species level, revealing specific patterns for the depressed and non-depressed groups without losing predictive power. We believe that this approach, and Machine Learning in general, can be really useful in a field of research were high dimensionality is always an issue.
This work provided, to our knowledge for the first time, an overview about the difference in the bacterial communities of patients with signs of depression and the combination with depression and inflammatory bowel disease. Our findings suggest a complex landscape of microbiome interactions, both at population structure and functional activity levels. However, the results showed that there are distinct taxonomic profiles within patients of IBD depending on their depression status, providing further input for future investigations.
The datasets used for the analyses were retrieved from the Inflammatory Bowel Disease Multi-Omics Database (IBDMDB) (Schirmer et al., 2018), a part of the Integrative Human Microbiome Project (NIH HMP Working Group et al., 2009).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (4)
I think part of the confusion might be phrases such as "running validation sets", or "validation models". If these refer to using the random forest models trained only on the training set to predict the disease labels on the test set, everything is fine. But people might understand a "validation model" to mean multiple things.
In the same vein, the article states "and among the considered models, only species that appeared more than once were selected". Does this mean that decision trees were pruned to only keep those that had these selected species as their initial steps? Or was a random forest re-trained using these selected species? In this case, either it was trained using the same training set used initially, or it was trained on the validation set.
I think part of the confusion might be phrases such as "running validation sets", or "validation models". If these refer to using the random forest models trained only on the training set to predict the disease labels on the test set, everything is fine. But people might understand a "validation model" to mean multiple things.
In the same vein, the article states "and among the considered models, only species that appeared more than once were selected". Does this mean that decision trees were pruned to only keep those that had these selected species as their initial steps? Or was a random forest re-trained using these selected species? In this case, either it was trained using the same training set used initially, or it was trained on the validation set.
The "validation set" was only used for prediction. The validation model was still trained with the "training set". We did this in order to see how ... Continue reading Dear Prof. Waldron,
The "validation set" was only used for prediction. The validation model was still trained with the "training set". We did this in order to see how our model fits new data and to avoid the scenario described in Cawley & Talbot, 2010, in section 5.3, where we are testing on samples that the model has already "seen". As you correctly point out, training on a different dataset would left us with no way to estimate the predictive power of our model.
The "validation set" was only used for prediction. The validation model was still trained with the "training set". We did this in order to see how our model fits new data and to avoid the scenario described in Cawley & Talbot, 2010, in section 5.3, where we are testing on samples that the model has already "seen". As you correctly point out, training on a different dataset would left us with no way to estimate the predictive power of our model.