Keywords
Transcription factors, Gene expression, Gene regulation, Bioconductor
This article is included in the Bioconductor gateway.
This article is included in the Bioinformatics gateway.
Transcription factors, Gene expression, Gene regulation, Bioconductor
The two reviewer reports raised valid concerns related to the clarity of presentation.
A reviewer has noted that the interpretation of figure 3 is difficult, and we concur. Additional work on the relationship between sequence-based and in vitro evidence of TF binding, specifically with respect to the combinatorial aspects of binding suggested by Figure 3, is warranted.
To demonstrate interplay of TFutils with existing Bioconductor tools, Figure 5 is new, and makes use of motifStack and MotifDb.
See the authors' detailed response to the review by Lihua Julie Zhu, Haibo Liu and Rui Li
See the authors' detailed response to the review by Giovanna Ambrosini and Philipp Bucher
See the authors' detailed response to the review by Kevin Ernst and Matthew T. Weirauch
A central concern of genome biology is improving understanding of gene transcription. In simple terms, transcription factors (TFs) are proteins that bind to DNA, typically near gene promoter regions. The role of TFs in gene expression variation is of great interest. Progress in deciphering genetic and epigenetic processes that affect TF abundance and function will be essential in clarifying and interpreting gene expression variation patterns and their effects on phenotype. Difficulties of identifying functional binding of TFs, and opportunities for using information of TF binding in systems biology contexts, are reviewed in Lambert et al.1 and Weirauch et al.2.
This paper describes an R/Bioconductor package called TFutils, which assembles various resources intended to clarify and unify approaches to working with TF concepts in bioinformatic analysis. Computations described in this paper can be carried out with Bioconductor version 3.8. The package can be installed with
# use install.packages("BiocManager") if not already available
library(BiocManager)
install("TFutils")In the next section we describe the basic concepts of enumerating and classifying TFs, enumerating TF targets, and representing genome-wide quantification of TF binding affinity. This is followed by a review of the key data structures and functions provided in the package, and an example in cancer informatics.
The present paper does not deal directly with the manipulation or interpretation of sequence motifs. An excellent Bioconductor package that synthesizes many approaches to these tasks is universalmotif.
A complete reference manual enumerating all functions and data sets in the package is available at: http://bioconductor.org/packages/release/bioc/manuals/TFutils/man/TFutils.pdf
Given the importance of the topic, it is not surprising that a number of bioinformatic research groups have published catalogs of transcription factors along with metadata about their features. Standard nomenclature for TFs has yet to be established. Gene symbols, motif sequences, and position-weight matrix catalog entries have all been used as TF identifiers.
In TFutils we have gathered information from four widely used resources, focusing specifically on human TFs: Gene Ontology (GO, Ashburner et al.3, in which GO:0003700 is the tag for the molecular function concept “DNA binding transcription factor activity”), CISBP (Catalog of Inferred Sequence Binding Preferences) (Weirauch et al.2), HOCOMOCO (Homo sapiens Comprehensive Model Collection) (Kulakovskiy et al.4), and the “c3 TFT (transcription factor target)” signature set of MSigDb (Molecular Signatures Database) (Subramanian et al.5). Figure 1 depicts the sizes of these catalogs, measured using counts of unique HGNC gene symbols. The enumeration for GO uses Bioconductor’s org.Hs.eg.db (version 3.7.0) package to find direct associations from GO:0003700 to HGNC symbols. The enumeration for MSigDb is heuristic and involves parsing the gene set identifiers used in MSigDb for exact or close matches to HGNC symbols. For CISBP and HOCOMOCO, the associated web servers provide easily parsed tabular catalogs.

As noted by Weirauch et al.2, interpretation of the “function and evolution of DNA sequences” is dependent on the analysis of sequence-specific DNA binding domains. These domains are dynamic and cell-type specific (Gertz et al.6). Classifying TFs according to features of the binding domain is an ongoing process of increasing intricacy. Figure 2 shows excerpts of hierarchies of terms related to TF type derived from GO (on the left) and TFclass (Wingender et al.7). There is a disagreement between our enumeration of TFs based on GO in Figure 1 and the 1919 shown in AmiGO, as the latter includes a broader collection of receptor activities.

Table 1 provides examples of frequently encountered TF classifications in the CISBP and HOCOMOCO catalogs. The numerical components of the HOCOMOCO classes correspond to TFClass subfamilies (Wingender et al.7).
The number of unique human TF_Name entries in CISBP is 1734. The number of unique Transcription factor entries in HOCOMOCO (Sept. 2018 version) is 678. Entries in columns Nc (Nh) are numbers of distinct TFs annotated to classes in columns CISBP (HO-COMOCO) respectively. Entries are ordered top to bottom by frequency of occurrence. There is no substantive correspondence between entries on a given row. Harmonization of class terminology is beyond the scope of this paper.
The Broad Institute MSigDb (Subramanian et al.5) includes a gene set collection devoted to cataloging TF targets. We have used Bioconductor’s GSEABase package (version 1.45.0) to import and serialize the gmt representation of this collection.
TFutils::tftColl ## GeneSetCollection ## names: AAANWWTGC_UNKNOWN, AAAYRNCTG_UNKNOWN, ..., GCCATNTTG_YY1_Q6 (615 total) ## unique identifiers: 4208, 481, ..., 56903 (12774 total) ## types in collection: ## geneIdType: EntrezIdentifier (1 total) ## collectionType: NullCollection (1 total)
Names of TFs for which target sets are assembled are encoded in a systematic way, with underscores separating substrings describing motifs, genes, and versions. Some peculiarity in nomenclature in the MSigDb labels can be observed:
grep("NFK", names(TFutils::tftColl), value=TRUE) ## [1] "NFKAPPAB65_01" "NFKAPPAB_01" "NFKB_Q6" ## [4] "NFKB_C" "NFKB_Q6_01" "GGGNNTTTCC_NFKB_Q6_01"
Manual curation will be needed to improve the precision with which MSigDb TF target sets can be associated with specific TFs or motifs.
In this subsection we address representation of putative binding sites. First we illustrate how to represent sequence-based affinity measures and the binding site locations implied by these. We then discuss use of results of ChIP-seq experiments for cell-type-specific binding site enumeration.
Affinity scores based on reference sequence. The FIMO algorithm of the MEME suite (Grant et al.8) was used to score the human reference genome for TF binding affinity for 689 motif matrices to which genes are associated. Full details of the execution of FIMO are provided in Sonawane et al9. Sixteen (16) tabix-indexed BED files are lodged in an AWS S3 bucket for illustration purposes.
library(GenomicFiles) data(fimo16) fimo16 ## GenomicFiles object with 0 ranges and 16 files: ## files: M0635_1.02sort.bed.gz, M3433_1.02sort.bed.gz, ..., M6159_1.02sort.bed.gz, M6497_1.02sort.bed. ## detail: use files(), rowRanges(), colData(), ...
head(colData(fimo16)) ## DataFrame with 6 rows and 2 columns ## Mtag HGNC ## <character> <character> ## 1 M0635_1 DMRTC2 ## 2 M3433_1 HOXA3 ## 3 M3467_1 IRF1 ## 4 M3675_1 POU2F1 ## 5 M3698_1 TP53 ## 6 M3966_1 STAT1
We harvest scores in a genomic interval of interest (bound to fimo16 in the rowRanges assignment below) using reduceByFile. This yields a list with one element per file. Each such element holds a list of scanTabix results, one per query range.
library(BiocParallel) register(SerialParam()) # important for macosx? rowRanges(fimo16) = GRanges("chr17", IRanges(38.077e6, 38.084e6)) rr = GenomicFiles::reduceByFile(fimo16, MAP=function(r,f) scanTabix(f, param=r))
scanTabix produces a list of vectors of text strings, which we parse with data.table::fread. The resulting tables are then reduced to a genomic location and -log10 of the p-value derived from the binding affinity statistic of FIMO in the vicinity of that location.
asdf = function(x) data.table::fread(paste0(x, collapse="\n"), header=FALSE) gg = lapply(rr, function(x) { tmp = asdf(x[[1]][[1]]) data.frame(loc=tmp$V2, score=-log10(tmp$V7)) }) for (i in 1:length(gg)) gg[[i]]$tf = colData(fimo16)[i,2]
It turns out there are too many distinct TFs to display names individually, so we label the scores with the names of the associated TF families as defined in CISBP.
matchcis = match(colData(fimo16)[,2], cisbpTFcat[,2]) famn = cisbpTFcat[matchcis,]$Family_Name for (i in 1:length(gg)) gg[[i]]$tffam = famn[i] nn = do.call(rbind, gg)
A simple display of predicted TF binding affinity near the gene ORMDL3 is provided in Figure 3.
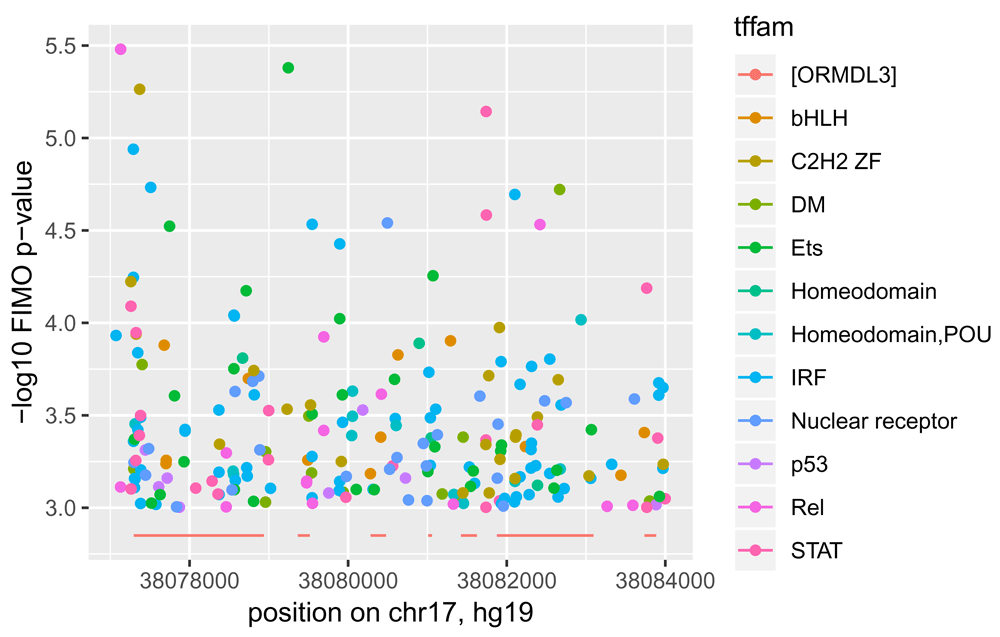
Points are -log10-transformed FIMO-based p-values colored according to TF class as annotated in CISBP. Segments at bottom of plot are transcribed regions of ORMDL3 according to UCSC gene models in build hg19.
TF binding predictions based on ChIP-seq data from ENCODE. The ENCODE project provides BED-formatted reports on ChIP-seq experiments for many combinations of cell type and DNA-binding factors. TFutils includes a table encode690 that gives information on 690 experiments involving pairs formed from 91 cell lines and 161 TFs for which results have been recorded as GRanges instances that can be acquired with the AnnotationHub (version 2.15.4) package. Positional relationships between cell-type specific binding sites and genomic features can be investigated. An illustration is given in Figure 4, in which is it suggested that in HepG2 cells, CEBPB exhibits a distinctive pattern of binding in the vicinity of ORMDL3.
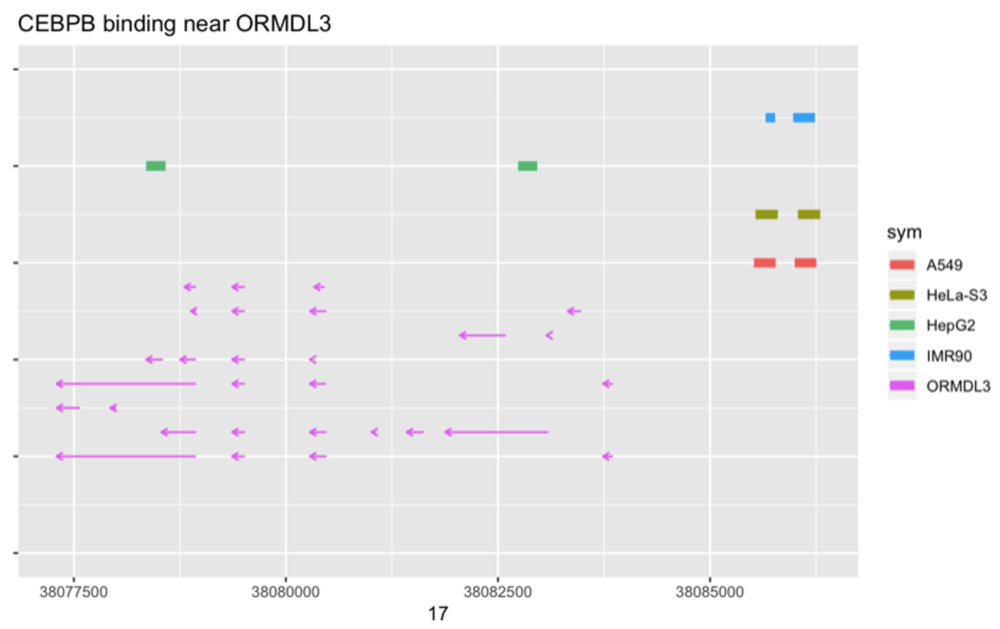
Colored rectangles at top are regions identified as narrow binding peaks, arrows in bottom half are exons in ORMDL3. Arrows sharing a common vertical position are members of the same transcript as cataloged in Ensembl version 75.
Inspired by a referee’s suggestion, we created functions that couple the HOCOMOCO TFclass enumeration with Bioconductor’s MotifDb10 and motifStack11 package resources. Figure 5 is the output of example(tffamCirc.plot), available in version 1.5.1 of TFutils.
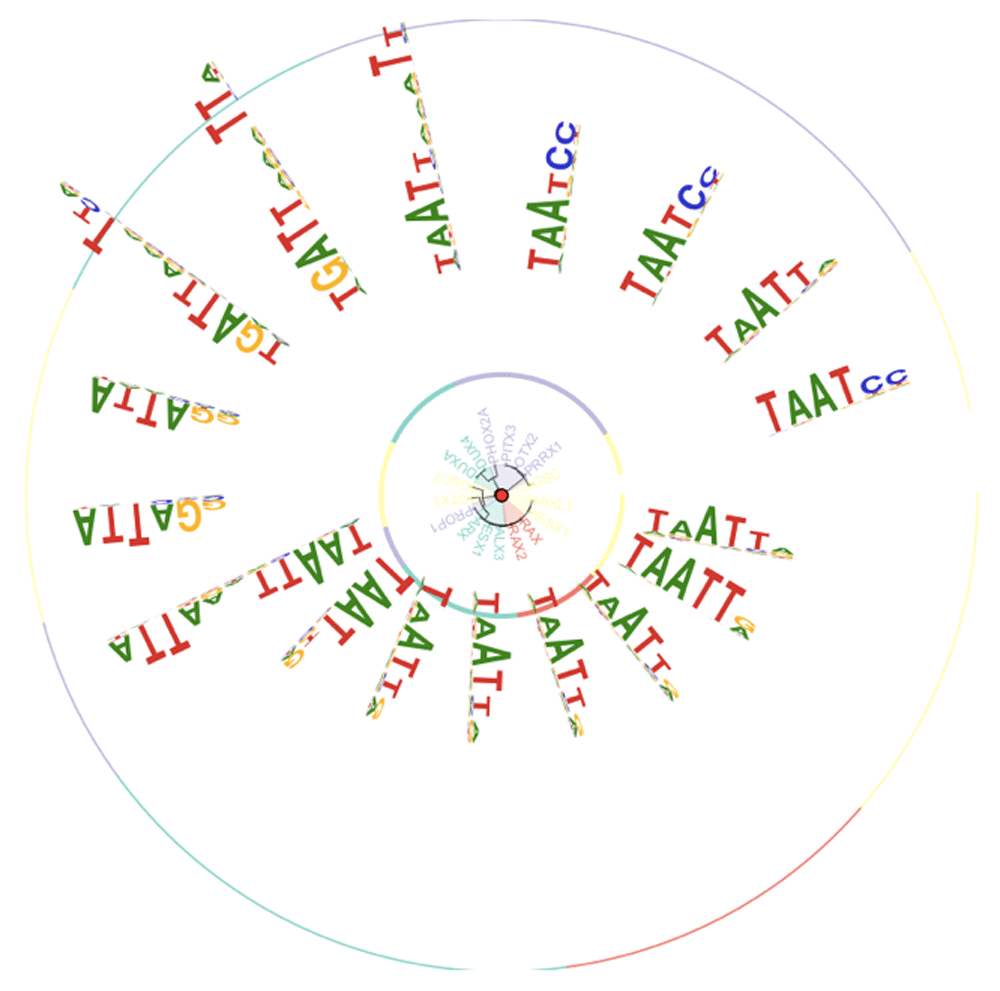
We have compared enumerations of human transcription factors by different projects, provided access to two forms of binding domain classification, and illustrated the use of cloud-resident genome-wide binding predictions. In the next section we review selected details of data structures and methods of the TFutils package.
The TFutils package is designed to lower barriers to usage of key findings of TF biology in human genome research. TFutils is supplied as a conventional R package distributed with, and making use of, the Bioconductor software ecosystem. TFutils includes ready-to-use reference data, tools for visualizing binding sites, and tools that simplify integrative use of TF binding information with GWAS findings. A complete enumeration of functions and data available in the package is provided in the reference manual at http://bioconductor.org/packages/release/bioc/manuals/TFutils/man/TFutils.pdf
Catalogs. Two reference resources have been collected into the TFutils package as data.frame instances. These are cisbpTFcat (CISBP: 7592 x 28), and hocomoco.mono.sep2018 (mononucleotide models, full catalog, 769 x 9). These data.frames are snapshots of the CISBP and HOCOMOCO catalogs.
Indexed BED in AWS S3. As described above fimo16 provides programmatic access to FIMO scores for 16 TFs, using the GenomicFiles (version 1.19.0) protocol.
Annotated reference to ENCODE ChIP-seq results. encode690 simplifies programmatic access to TF:cell-line combinations available in Bioconductor AnnotationHub (version 2.15.4).
TF targets enumerated in MsigDb. The c3-TFT (TF targets) subset from MSigDb is provided as a GeneSet-Collection instance as defined in GSEABase.
Illustrative GWAS records. The full EBI/EMBL GWAS catalog is available in the gwascat package (version 2.15.0); for convenience, an excerpt focusing on chromosome 17 is supplied with TFutils as gwascat_hg19_chr17.
Interactive enumeration of TF targets implicated in GWAS. The TFtargs function runs a shiny app that permits selection of a TF in the nomenclature of the MSigDb c3/TFT gene set collection. The app will search an object provided by the gwascat package for references in the MAPPED_GENE field that match the targets of the selected TF. Figure 6 gives an illustration.

This example reports on recent EBI GWAS catalog hits on chromosome 17 only.
The TFCatalog S4 class. Reference catalogs for TF biology are structured with the TFCatalog S4 class. Two essential components for managing a catalog are the native TF identifier for the catalog and the HGNC gene symbol typically used to name the TF. The TFCatalog class includes a name field to name the catalog, and a character vector with elements comprised of the native identifiers for catalogued TFs.
For example, CISBP uses T004843_1.02 to refer to motifs associated with gene TFAP2B. There are five such motifs, three derived from SELEX, one from Transfac, and one from Hocomoco.
A data.frame instance that has an obligatory column named ‘HGNC’ can include any collection of fields that offer metadata about the TF in the specified catalog. Here is how we construct and view a TFCatalog object using the CISBP reference data.
data(cisbpTFcat) TFs_CISBP = TFCatalog(name="CISBP.info", nativeIds=cisbpTFcat[,1], HGNCmap = cisbpTFcat) TFs_CISBP ## TFutils TFCatalog instance CISBP.info ## 7592 native Ids, including ## T004843_1.02 ... T153733_1.02 ## 1551 unique HGNC tags, including ## TFAP2B TFAP2B ... ZNF10 ZNF350
The TFutils package can be installed in any version of R subsequent to 3.5.0, and therefore will be usable on Unix, Windows, or Mac platforms. The preferred method of installation employs the CRAN package BiocManager, through the R command BiocManager::install("TFutils"). All necessary dependencies will be installed through this process.
In this section we consider applications of the tools in genetic epidemiology. First we look for TFs that may harbor variants associated with traits in the EBI GWAS catalog. Then we show how to enumerate traits associated with targets of a selected TF.
Find TFs that are direct GWAS hits for a given trait. directHitsInCISBP accepts a string naming a trait, and returns a data.frame of TFs identified as “mapped genes” for the trait, with their TF “family name”.
library(dplyr) library(magrittr) library(gwascat) data(ebicat37) directHitsInCISBP("Rheumatoid arthritis", ebicat37) ## Joining, by = "HGNC" ## HGNC Family_Name ## 1 ARID5B ARID/BRIGHT ## 7 EOMES T-box ## 15 GATA3 GATA ## 35 JAZF1 C2H2 ZF ## 37 MECP2 MBD ## 45 MTF1 C2H2 ZF ## 57 REL Rel ## 65 STAT4 STAT ## 79 AIRE SAND ## 82 IRF5 IRF
Retrieve traits mapped to genes that are targets of a given TF. topTraitsOfTargets will acquire the targets of a selected TF, check for hits in these genes in a given GWAS catalog instance, and tabulate the most commonly reported traits.
tt = topTraitsOfTargets("MTF1", TFutils::tftColl, ebicat37) ## remapping identifiers of input GeneSetCollection to Symbol... ## done head(tt) ## DISEASE.TRAIT MAPPED_GENE SNPS CHR_ID ## 1 Atopic dermatitis TNXB rs41268896 6 ## 2 Atopic dermatitis TNXB rs12153855 6 ## 3 Atopic dermatitis KIF3A rs2897442 5 ## 4 Attention deficit hyperactivity disorder SEMA3A rs797820 7 ## 5 Attention deficit hyperactivity disorder DNM1 rs2502731 9 ## 6 Attention deficit hyperactivity disorder GPC6 rs7995215 13 ## CHR_POS ## 1 32102292 ## 2 32107027 ## 3 132713335 ## 4 83979723 ## 5 128214278 ## 6 93756253 table(tt[,1]) ## ## Atopic dermatitis ## 3 ## Attention deficit hyperactivity disorder ## 3 ## Height ## 7 ## Menarche (age at onset) ## 4 ## Obesity-related traits ## 11 ## Rheumatoid arthritis ## 3
Sources and consequences of variations in DNA transcription are fundamental problems for cell biology, and the projects we have made use of for cataloging transcription factors are at the boundaries of current knowledge.
It is noteworthy that the four resources used for Figure 1 agree on names of only 119 TFs. The fact that CISBP distinguishes 475 TFs that are not identified in any other source should be better understood. We observe that the ascription of TF status to AHRR is based on its sharing motifs with AHR (see http://cisbp.ccbr. utoronto.ca/TFreport.php?searchTF=T014165_1.02).
Figure 2 and Table 1 show that the classification of TFs is now fairly elaborate. Use of the precise terminology of the TFClass system to label TFs of interest at present relies on associations provided with the HOCOMOCO catalog.
As population studies in genomic and genetic epidemiology grow in size and scope, principles for organizing and prioritizing loci associated with phenotypes of interest are urgently needed. Figure 6 shows that loci associated with phenotypes related to kidney function, lung function, and IL-8 levels are potentially unified through the fact that the GWAS hits are connected with genes identified as targets of VDR (vitamin D receptor). This example limited attention to hits on chromosome 17; the TFtargs tool permits ad libitum exploration of phenotype-locus-gene-TF associations. Our hope is that the tools and resources collected in TFutils will foster systematic development of evidence-based mechanistic network models for transcription regulation in human disease contexts, thereby contributing to the development of personalized genomic medicine.
With the exception of the FIMO scoring data (fimo16), all data underlying the results are available as part of the article and no additional source data are required.
fimo16 links to indexed bed files in a public S3 bucket funded by the Bioconductor foundation. The underling data is sourced from Sonawane et al. 2017 https://doi.org/10.1016/j.celrep.2017.10.0019
Source code is available from GitHub: https://github.com/vjcitn/TFutils
Archived source code: https://doi.org/doi:10.18129/B9.bioc.TFutils12
Licence: Artistic License 2.0
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)