Keywords
Metastatic breast cancer, carboplatin, gemcitabine, chemotherapy
This article is included in the Oncology gateway.
This article is included in the All trials matter collection.
Metastatic breast cancer, carboplatin, gemcitabine, chemotherapy
Breast cancer is the most prevalent cancer in women, with over 1.5 million cases diagnosed annually worldwide1. Of these patients, 20 to 50% will develop metastatic disease2, with which only 25% will survive to 5 years3. Anthracycline- or taxane-based regimens form the mainstay of chemotherapy treatment in metastatic breast cancer; however, after failure of these first-line therapies, management of progressive disease is difficult and response rates for salvage therapies are as low as 25%4–6. Hence, further investigation into alternative chemotherapy regimens is required.
Gemcitabine is a pyrimidine analogue and antimetabolite drug with proven anti-tumour activity and tolerability in metastatic breast cancer7. In addition, gemcitabine is an excellent agent in polychemotherapy, as demonstrated by its potential for synergistic interactions with platinum agents8,9. Gemcitabine has been used in combination with platinum agents extensively in non-small cell lung cancer (NSCLC) with good results10 and, in this combination, carboplatin appears to be better tolerated than cisplatin, with similar efficacy rates11.
We proposed a prospective phase II trial to assess the efficacy of gemcitabine in combination with carboplatin in patients with human epidermal growth factor 2 (HER-2) negative, locally advanced or metastatic breast cancer that were anthracycline- and taxane-resistant.
Patients with locally advanced or metastatic breast cancer that had previously been treated with anthracyclines and taxanes were eligible to be screened for entry into the study. The protocol was approved by an independent research ethics committee and all patients gave written informed consent prior to enrolment. Research was conducted in accordance with the Good Clinical Practice guidelines. Consent materials and the full protocol are available as Extended data12.
Patients were approached opportunistically within the hospital setting and screened for eligibility by hospital research staff to ensure all inclusion and exclusion criteria were met. Clinicians sought informed consent to enter the trial from a patient only after the patient had received a full explanation of the trial, had read the PIS and had enough time to consider taking part. Data were collected at the investigational site.
Patients were eligible if they had histologically proven HER-2-negative breast cancer and a diagnosis of locally advanced or metastatic breast cancer, and had previously received treatment with an anthracycline and a taxane either as neoadjuvant, adjuvant or metastatic therapy. Patients were not eligible if they had received more than one prior course of chemotherapy for metastatic disease. Prior hormonal or immunotherapy was allowed, provided anti-tumoral hormonal therapy was terminated prior to enrolment. Prior radiotherapy was also allowed, providing <25% of the bone marrow was treated, the radiotherapy was completed >4 weeks prior to enrolment, the whole pelvis was not irradiated and the patient had recovered from the acute toxic effects. Irradiated lesions were not included as sites of measurable disease.
Patients were required to have at least one measurable site of disease, defined as a lesion that could be accurately measured in at least one dimension as 2 cm or greater with conventional techniques or as 1 cm or greater with spiral CT scan. Palpable disease was acceptable. Where there was a solitary site of recurrence, histological or cytological confirmation was required.
Other eligibility criteria included: age ≥18 years; Eastern Cooperative Oncology Group (ECOG) performance score ≤1; minimal life expectancy of 12 weeks; adequate bone marrow reserve; adequate renal and hepatic function.
Patients were not eligible for enrolment if they: were pregnant, breast-feeding or refused approved contraception if of reproductive potential; had serious medical or psychiatric illness or serious active infection; had a history of a second primary malignancy other than cervical carcinoma in situ, nonmelanomatous carcinoma of the skin or other malignancy treated at least 5 years previously with no evidence of recurrence; had NCIC Common Toxicity Criteria (CTC) grade 2 peripheral neuropathy; or intended to start or stop bisphosphonate therapy within 4 weeks of enrolment. Prior administration of gemcitabine, cisplatin or carboplatin, or concomitant anti-cancer treatment was not permitted. Additionally, patients who received cytotoxic chemotherapy within the preceding 21 days or a drug without regulatory approval within the preceding 30 days prior to enrolment were not eligible.
Within the 2 weeks prior to commencing treatment, pre-trial investigations were performed. These included taking haematological samples for full blood count, serum biochemistry, hepatic function and estimation of glomerular filtration rate by Cockcroft Gault calculation, and performing a computed tomography (CT) scan of the chest, abdomen and pelvis.
Patients were reviewed regularly throughout treatment. Prior to each 2-week cycle, haematological samples were obtained for full blood count, serum biochemistry and estimation of glomerular filtration rate by Cockcroft Gault calculation. In addition, patients were also required to undergo a physical examination and complete an ECOG performance questionnaire and toxicity assessment.
To assess tumour response, CT scanning of the chest, abdomen and pelvis was performed after cycles 3, 6 and 9 (where applicable). The longest diameters of target lesions were recorded (maximum of five lesions per organ and 10 lesions in total). Lesions were selected on basis of their size (lesions with the longest diameter) and suitability for accurate repeated measurements. Response assessment was based on the Response Evaluation Criteria in Solid Tumours (RECIST) criteria and classified as a complete response (disappearance of all target lesions), partial response (at least 30% decrease in the sum of the longest diameter of target lesions), progressive disease (at least 20% increase in the sum of the longest diameter of target lesions or the appearance of one or more new lesions) or stable disease (neither sufficient shrinkage to qualify for partial response nor sufficient increase to qualify for progressive disease). Overall response rate was defined as the proportion of patients exhibiting a complete or partial response at the end of treatment.
Patients were followed up to obtain data on overall survival (time from date of study entry to date of death from any cause), time to progression (time from date of study entry to date of documented progression or death from any cause), duration of response (time from first documented tumour response to the date of disease progression) and time to treatment failure (time from date of study entry to the date of premature discontinuation of study treatment for any reason, or death from any cause, whichever was sooner).
All patients received intravenous carboplatin (dissolved in 250ml 5% dextrose) at a dose consistent with a target area under curve (AUC)13 of 4.5 mg/ml.min given over 30 minutes on day 1, and intravenous gemcitabine 1500 mg/m2 (up to a maximum dose of 2000 mg) at a fixed dose rate (FDR) of 10 mg/m2/min on day 2 of every 2-week cycle. This biweekly schedule has been shown to be possible in NSCLC14, where a FDR of gemcitabine15,16 and preceding gemcitabine dosing with carboplatin17 both seem to result in greater efficacy. Body surface area was calculated on each patient’s actual height and weight at the commencement of treatment.
The target number of treatment cycles was six. Patients received three treatment cycles initially, followed by a CT scan of the chest abdomen and pelvis. Patients without disease progression then received three further treatment cycles. Patients with an ongoing response as demonstrated by a further CT scan after cycle 6 were eligible to be offered an additional three cycles at the investigators’ discretion. Study treatment could be discontinued at any point due to: patient request; recurrent grade 3 or 4 treatment-related toxicity despite dose reduction; allergic reaction to a study drug; evidence of disease progression after two or more cycles of treatment; if the patient became pregnant; or if the responsible clinician felt it was not in the patients best interests to continue on study treatment.
Doses were modified based on absolute neutrophil counts (ANC), platelet counts and non-haematological toxicity assessments prior to each cycle. Doses of both gemcitabine and carboplatin were reduced to 80% of the starting dose when ANC decreased to <1000/mm3 or after a first episode of uncomplicated febrile neutropenia. After a first episode of febrile neutropenia with hypotension or requiring hospital admission for more than 4 days, or a second episode of uncomplicated febrile neutropenia, doses were reduced to 60% of the starting dose or treatment was stopped if there was no evidence of response or the patient was not fit for further chemotherapy. Treatment was discontinued after a third episode of uncomplicated febrile neutropenia, or after a second episode of febrile neutropenia with hypotension or requiring hospital admission for more than 4 days.
When platelet counts decreased to <100,000/mm3, cycles were delayed until platelets recovered (>100,000/mm3). After a second episode of thrombocytopenia (<100,000/mm3), doses of both gemcitabine and carboplatin were reduced to 80% of the starting dose. The dose was further reduced to 60% of the starting dose after a third episode. After a first episode of grade 4 thrombocytopenia or thrombocytopenia-related bleeding, doses of both gemcitabine and carboplatin were reduced to 80% of the starting dose; and this was further reduced to 60% of the starting dose after a second episode.
Clinical assessment of non-haematological toxicity was performed prior to each cycle. Doses were reduced to 80% of the starting dose for CTC grade 3 or 4 toxicities, or to 60% of the starting dose for CTC grade 4 toxicities. In patients with pneumonitis grade 2 or greater, gemcitabine was discontinued. Treatments could also be delayed by up to 2 weeks for any reason deemed appropriate for the treating clinician.
The primary endpoint was to determine the overall response rate. Secondary endpoints included overall toxicity, overall survival, time to disease progression, duration of response and time to treatment failure.
A two-stage sampling design was employed18. For the purposes of this study, a response rate of less than 10% (null hypothesis) was considered to yield no advantage over existing therapies. Conversely, a response rate of 30% or greater (alternative hypothesis) was to indicate that the experimental regimen was of interest and warranted further evaluation. In order to reach a power of 0.9, the recruitment target was 35 patients.
The trial was entered into EudraCT on 10-APR-2006 (EudraCT Number: 2005-005164-83); https://www.clinicaltrialsregister.eu/ctr-search/trial/2005-005164-83/GB.
The trial received ethical approval from Southampton & South West Hampshire REC (A) (06/Q1702/46) on 15 June 2006. It received approval from the UK Medicines and Health Care Product Regulatory Agency (MHRA) to be conducted in the UK (MHRA CTA number 11709/0208/001-0002). Southampton Clinical Trials Unit (SCTU), a UK Clinical Research Collaboration-registered CTU, coordinated the trial. Southampton University Hospitals NHS Trust was the sponsor for the trial (email: R&[email protected]).
In total, five patients were recruited from two secondary care sites in the United Kingdom between 2007 and 2008. The study was terminated early due to difficulty in accruing patients in November 2008.
Of the 34 patients screened, 29 (85%) did not meet the entry criteria and only two of the six participating sites were able to recruit patients. The most common reasons for screen failures included patients having had no previous adjuvant chemotherapy (31%) and HER-2 positivity (17%), as shown in Figure 1. Delays were also experienced waiting for local Research and Development departments to approve the study and not returning essential documents in a timely manner. The Trial Steering Committee reviewed the data and concluded that the trial would not complete in a reasonable timeframe. The primary endpoint was evaluated in all patients.
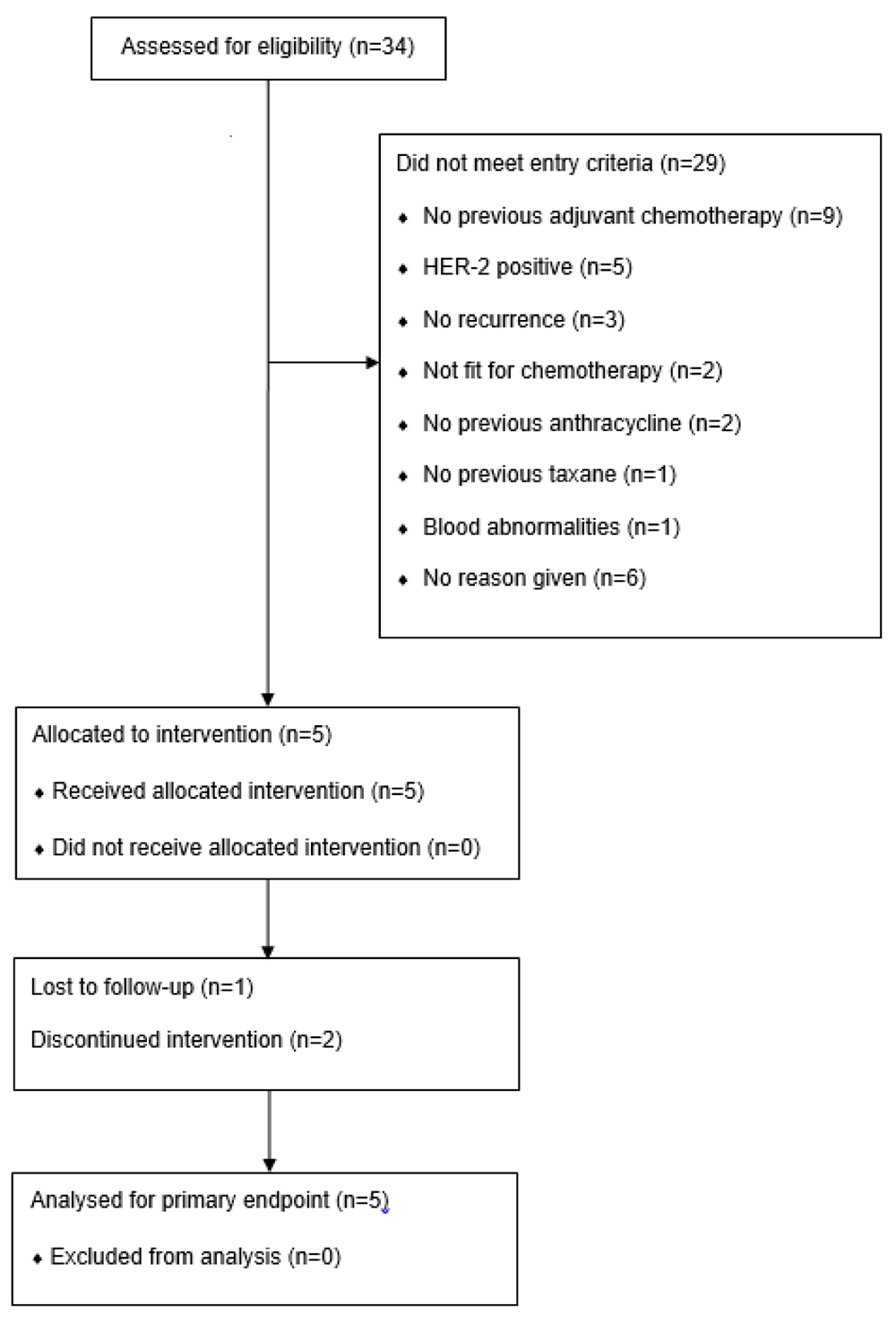
The median age of participants was 38 years (range 30 to 72 years), and all five patients were female. Patient characteristics at study entry are shown in Table 1.
| Variable | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 |
|---|---|---|---|---|---|
| Age, years | 38 | 46 | 72 | 30 | 35 |
| Sex | Female | Female | Female | Female | Female |
| ECOG PS | 1 | 1 | 0 | 0 | 0 |
| Histological type | Ductal adenocarcinoma | Ductal | Mammary adenocarcinoma | Ductal adenocarcinoma | High grade DCIS |
| Baseline target lesions | 6 | 1 | 1 | 3 | 5 |
| Baseline non-target lesions | 4 | 4 | 1 | 1 | 0 |
| Sites of metastatic disease | Lung, lymph node, liver | Liver, lung | Lymph node, lung | Lung | Liver |
| Oestrogen receptor status | Negative | Positive | Negative | Negative | Negative |
| Type of neoadjuvant chemotherapy | - | - | - | - | Doxorubicin, cyclophosphamide, epirubicin |
| Type of adjuvant chemotherapy | Epirubicin + CMF | NEAT trial chemotherapy19 | FEC | FEC, Docetaxel | Docetaxel |
| Type of metastatic chemotherapy | Docetaxel | Docetaxel | Docetaxel | - | - |
| Prior radiotherapy | Yes | Yes | Yes | Yes | Yes |
| Prior hormonal treatment | Goserelin | Tamoxifen | No | No | Tamoxifen, Goserelin |
Gemcitabine and carboplatin were administered intravenously by the Investigator or member of their clinical team to individual patients at the investigational sites. As a result, patient compliance monitoring was ensured. Patients who returned for follow up visits received study drug unless they encountered toxicity problems or their disease had progressed.
The study case report form was monitored by Southampton Clinical Trials Unit trial staff to ensure the gemcitabine and carboplatin doses were administered as scheduled.
The median number of cycles received was six (range 3 to 9 cycles). Two patients received less than 6 cycles, one due to progressive disease and one due to toxicity. One patient received 9 cycles due to a complete response.
Of the received cycles, six doses (21%) of 28 doses of gemcitabine planned were reduced, and 15 doses (54%) delayed. No doses were omitted. The reason for reduction was myelosupression (thrombocytopenia and anaemia), and the main reason for delay was neutropenia. Likewise, six (21%) of the 28 doses of carboplatin planned were reduced, and 15 doses (54%) delayed. No doses were omitted. The reason for reduction was myelosupression (anaemia and neutropenia), and the main reason for delay was neutropenia.
Notably, in three of five of the patients, the protocol was incorrectly followed by the investigating site and, in response to neutropenia, doses of both gemcitabine and carboplatin were delayed rather than reduced.
Of the five patients who completed treatment, one patient achieved a complete response, one patient achieved a partial response, one patient had stable disease and two patients had disease progression. The patient with a complete response was lost to follow-up. All four of the remaining patients died, and the median overall survival was 6.3 months (range 5.0–31.6). The median time to treatment failure was 3.9 months (range 1.8–5.4), and the median time to progression was 4.7 months (range 1.8–31.6). For the patient with a partial response, the duration of response was 4.1 months.
All patients were evaluated for toxicity (Table 2). The predominant toxicities which occurred in all patients were neutropenia and anaemia. One patient developed neutropenic sepsis and subsequently discontinued treatment. There were no severe non-haematological toxicities.
In this study, gemcitabine in combination with carboplatin seemed to be effective in treating a small number of HER-2-negative advanced or metastatic breast cancer patients. The study, however, closed early and did not reach the recruitment target of 35 patients to provide adequate power for statistical analysis. Hence, we are unable to draw firm conclusions regarding the efficacy of this combination from this study.
The major reason for early closure was difficulty in recruitment. It proved difficult to source patients who had had previous chemotherapy, including an anthracycline and a taxane, and who were HER-2 negative and fit to receive further chemotherapy. The entry criteria proved to be too stringent, limiting the number of eligible patients. This is important for future studies to take into consideration because, if using similar criteria, recruitment targets may not be met within a reasonable timeframe.
The response rate seen in this study was 40%. In view of the young study population who had not had a large amount of pre-treatment, a reasonable response rate was expected. There are now several other small phase II studies that have tested the combination of gemcitabine and carboplatin in pre-treated metastatic breast cancer20–24. These trials yielded lower response rates of 30–39%, with times to progression of 4.6–7.0 months. Entry criteria were generally less stringent, for example not requiring patients to be pre-treated with an anthracycline and taxane23–25 or including patients with any HER-2 status22–24, which may account for success in accruing patients to these studies.
Significant haematological toxicities were seen in this trial, with all patients developing neutropenia and anaemia, 60% and 40% of which were grade 3/4 respectively. One patient developed grade 4 febrile neutropenia. The proportion of patients developing haematological toxicities was comparable to other phase II studies testing the combination, where febrile neutropenia occurred in 10–15% of patients, grade 3/4 neutropenia in 10–58%, grade 3/4 thrombocytopenia in 9–51% and grade 3/4 anaemia in 10–27%20–24. Non-haematological toxicities were not severe.
In contrast to other phase II studies where gemcitabine has been given at 1000mg/m2 on day 1 and 8 of every 3 week cycle20–24, we gave 1500mg/m2 on day 2 of every 2 week cycle at a FDR of 10mg/m2/min, after it was suggested this may be more efficacious in NSCLC15,16. Due to the failure to meet recruitment targets and deviations from the protocol at one centre, further investigation is required to determine the efficacy of this regimen in metastatic breast cancer.
In summary, this study highlights the difficulty in accruing HER-2 negative metastatic breast cancer patients who have been pre-treated with both anthracyclines and taxanes. In addition, although firm conclusions regarding efficacy cannot be made due to insufficient power, this study suggests the combination of gemcitabine and carboplatin is able to be delivered to this patient population and may be efficacious, in line with other phase II studies which have now been conducted.
Pseudonymised individual participant data (IPD) within the clinical trial dataset are available for sharing via controlled access by authorised SCTU staff (as delegated to SCTU by the trial sponsor). Data access can be requested via a SCTU Data Release application form (available from https://www.southampton.ac.uk/ctu/about/index.page); detailing the specific requirements and the proposed research, statistical analysis, publication plan and evidence of research group qualifications. Please email the completed form to the SCTU Data Release Committee Coordinator at [email protected].
Data access requests are reviewed against specific eligibility criteria by the SCTU data custodian and key members of the trial team, including a statistician and chief investigator or by an external Independent Review Panel. Decisions about requests are made promptly and usually no more than three months after receipt of request. Responses to all data requests, with a clear rationale for any refusals, will be sent promptly to the data requester.
Figshare: A phase 2 open-label study of carboplatin in combination with gemcitabine as a dose-dense schedule in patients with locally advanced or metastatic breast cancer that are resistant to anthracyclines and taxanes. https://doi.org/10.6084/m9.figshare.11417550.v112.
The following extended data are available:
Figshare: TREND Statement for ‘A phase 2 open-label study of carboplatin in combination with gemcitabine as a dose-dense schedule in patients with locally advanced or metastatic breast cancer that are resistant to anthracyclines and taxanes’. https://doi.org/10.6084/m9.figshare.11417550.v112.
Extended data and reporting guidelines are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)