Keywords
Mutational Signatures, pmsignature, COSMIC, Web interface, Shiny, R
This article is included in the RPackage gateway.
Mutational Signatures, pmsignature, COSMIC, Web interface, Shiny, R
Each human is subject to a variety of mutational processes throughout their lifetime, which result in the formation of a catalog of somatic mutations as his/her unique mutational profile1. A mutational signature captures the pattern of the mutations and contexts in which those mutations occur (i.e., the neighboring bases). Examples of important mutational processes with distinct mutational signatures include aging and ultraviolet (UV) radiation. Additionally, many research groups are performing analysis to discover de novo mutational signatures in cancer1–4.
Currently, there are two frameworks used to characterize and visualize mutational signatures5,6. The first, proposed by Alexandrov et al., used a vector of 96 probabilities to capture the possible composition of the six nucleotide substitutions (C>A, C>T, C>G, T>A, T>C, T>G) and the neighboring base immediately on each of the 5′ and 3′ side of the mutated base1. A list of published mutational signatures can be downloaded from the Catalogue Of Somatic Mutations In Cancer (COSMIC) website7 (version 2, v2). Later, Alexandrov et al. published an expanded set of mutational signatures, which was referred to as version 3 (v3)8. The 67 COSMIC v3 Single Base Substitution (SBS) signatures include 30 v2 signatures. Based on the signature concept, but using different model assumptions, Shiraishi et al. proposed a mixed-membership model, pmsignature, which substantially reduced the number of parameters needed to characterize a signature9. They achieved this by assuming independence across bases, thereby reducing the number of parameters from 6*4*4-1 = 95 to (6-1)+(4-1)+(4-1) = 119. The reduction in the number of parameters is greater if more flanking bases are included. All Shiraishi’s signatures can be downloaded from their GitHub repository9. In this paper, we will refer to signatures resulting from these two methods as “COSMIC signatures” with version numbers (for those resulting from Alexandrov et al.’s method) and “PM signatures” (for those resulting from Shiraishi et al.’s method).
A large number of researchers have published scientific findings resulting from the COSMIC signature-based method10–12, which was defined as the “gold standard" in the field by Baez-Ortega et al.6. Meanwhile, an increasing number of researchers are using the pmsignature-based method for samples with lower numbers of somatic variants due to it requiring fewer parameters9,13,14. Given that both methods are widely used, investigators need the ability to compare results from their analysis with those reported in earlier databases, which may have been produced using the alternate method. For example, researchers have adopted both tools for gastric cancer and tried to compare and integrate the information from two data sources in a somewhat ad hoc manner15. No rigorous tool exists for this task. In this paper we present iMutSig, an easy-to-use tool that allows users to identify the most similar mutational signatures across methods and to compare the information characterizing those signatures using simple point-and-click navigation.
In order to measure the similarity between mutational signatures across two databases, we need to represent PM signatures in a way that is comparable with those from COSMIC. To do this we convert the PM signature into a probabilistic vector with the same length as the COSMIC signature, i.e., 96. To calculate each of 96 resulting probabilities in the vector, we take the constituent components that make up the COSMIC signature - which refer to the nucleotide substitution and two flanking bases at the -1 and +1 position - calculate the probability of each component for the given PM signature, and then multiply those probabilities using pm-signature’s assumption of independence. For example, to calculate the probability of the COSMIC signature C[C >A]T we multiply three PM signature’s probabilities: P(C at pos -1), P(C >A), and P(T at pos +1). This example is shown in Table 1 and Equation 1.
Now that we have represented both forms of signature using probabilistic vectors of length n = 96, P and C say, we can directly compare the two signature types. In order to measure the similarity between them we use cosine similarity, CS, defined as shown in Equation 2:
Intuitively speaking, cosine similarity is the cosine of the angle between the two vectors. As such, cosine similarity ranges from 0 to 1 (inclusive). In our context, if two mutational signatures have a cosine similarity of 1, they must be identical, i.e., the angle between them is 0°; in contrast, if two mutational signatures have a cosine similarity of 0, they are maximally dissimilar (i.e., orthogonal). Computing the cosine similarity between the input signature and each of the candidate signatures, and then sorting the similarities from highest to lowest value, we identify the candidate signature with the highest cosine similarity as the most similar mutational signature.
iMutSig is built in R with its key features depending on the R package, pmsignature9. As shown in Figure 1, the Shiny app currently supports three possible workflows for users to choose from, depending on the type of signatures they have already obtained: 1) starting with a COSMIC signature; 2) starting with a PM signature; 3) starting with a self-defined signature that could follow either the COSMIC or PM format.
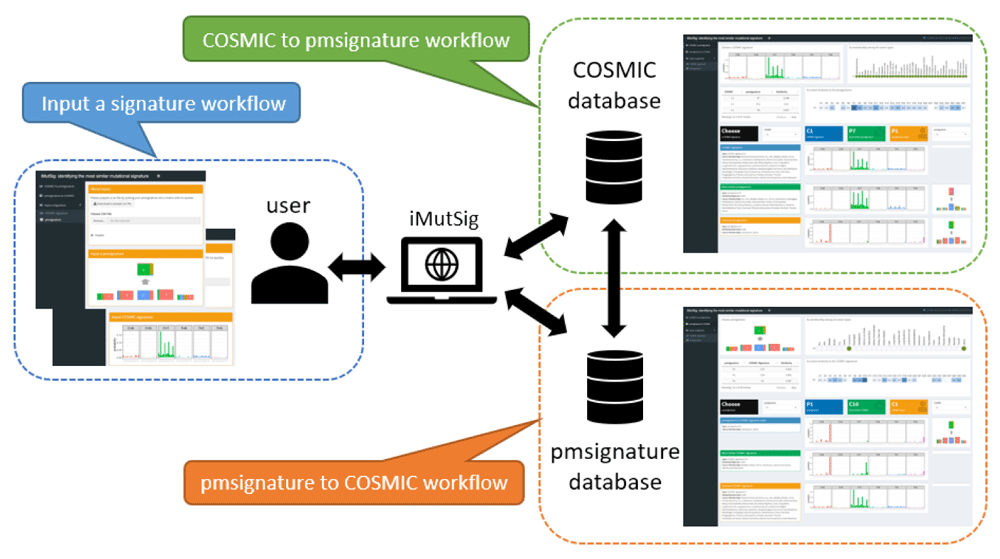
The first two tabs allow users to finding the most similar PM signature to an input COSMIC signature (highlighted in green) and vice versa (highlighted in orange). In addition, users can identify the most similar signatures from both data sources to an input signature (highlighted in blue).
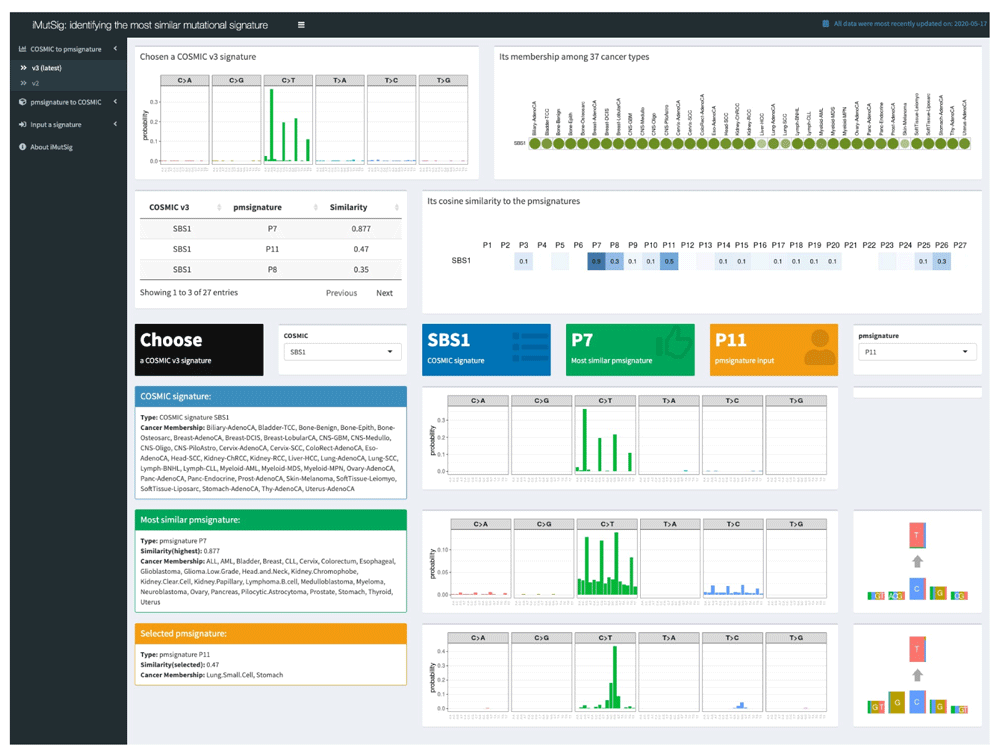
The first tab in the Shiny app window, “COSMIC to pmsignature", allows users to select an input COSMIC signature via a drop-down list and returns the best-matched PM signature. The returned results are divided and organized separately in the top and the bottom portion of the page. The top half tab summarizes background information regarding the input signature by presenting: 1) visualized plots of the input signature and its membership among all cancer types, i.e., in which kind of cancers the mutational signatures has been found; 2) a table showing the cosine similarity between this signature and all PM signatures, sorted in decreasing order, along with a visualization of a similarity heatmap with color and intensity proportional to assessed similarity. The bottom half tab presents plots and descriptions of the input COSMIC signature, the most similar PM signature, and a second PM signature that the user can select. Thus, users can easily access all the vital information and results regarding these signatures rather than having to manually gather and organize information from publications. The top half of the tab will be automatically updated via a control panel in the middle section of the tab, which enables users to select a signature to start with and also highlights information about the currently selected signature, the most-similar signature from the alternate model framework, and the cosine similarity.
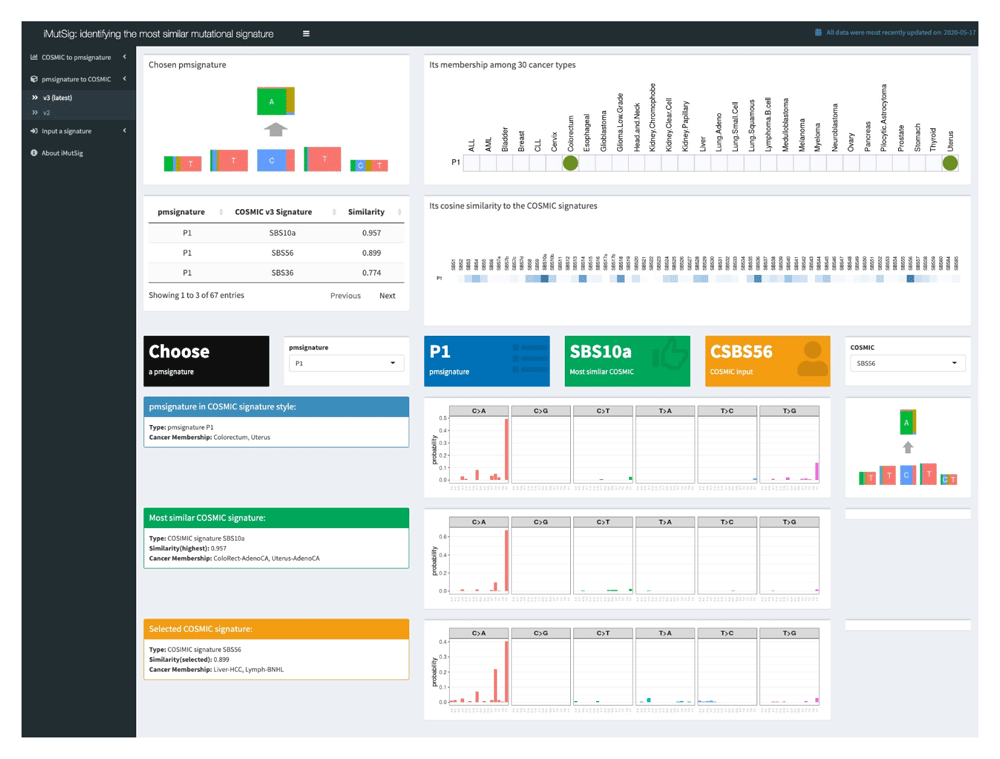
The second tab was designed in a similar manner to the first tab, but for the case in which we are starting with a PM signature and looking for the most similar COSMIC signature. For the first two tabs, users can choose which version of COSMIC signatures to input from the sub-menus, i.e., v2 or v3.
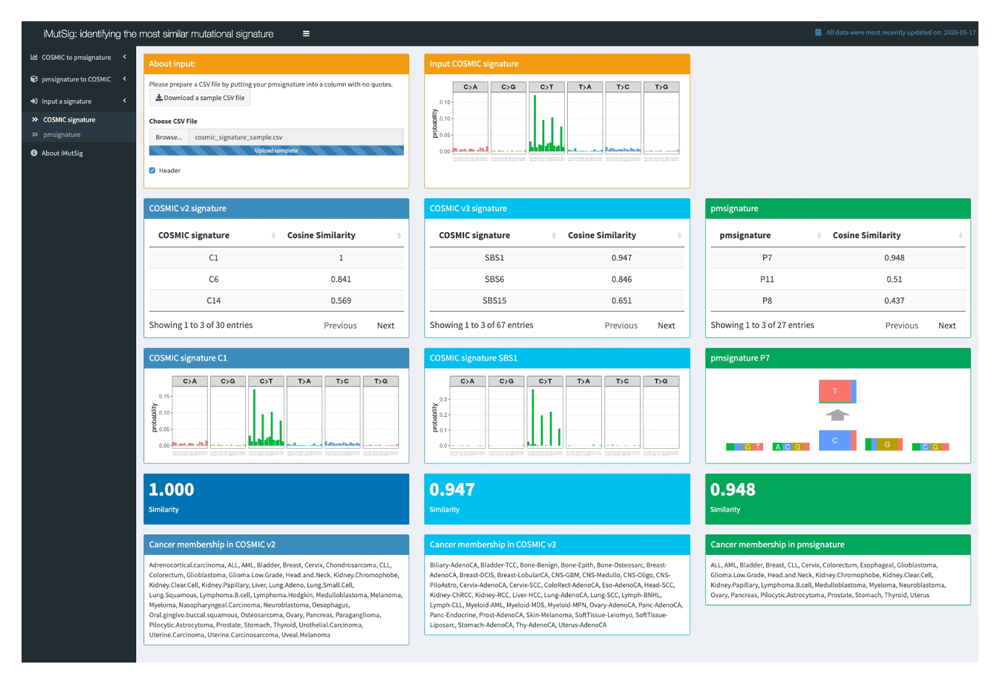
Unlike the first two tabs, the third tab enables users to enter a user-supplied signature, which can be in either PM or COSMIC format, and then identify the most similar signature from each online database. The user will be requested to enter a sub-menu based on the type of the input signature and to upload a comma-separated values (CSV) file containing a single signature. A sample CSV file is provided for download to give the user a better sense of the format of the input file. Then, the tab will be updated to display three tables, one from each data source (COSMIC v2, v3 and PM), listing the signatures from that data source and the cosine similarity of each signature with the user-uploaded signature. The tables are ordered from most similar to least similar signature. In addition, the user is able to view figures of the best-matched signatures (i.e., those with highest cosine similarity) from each data source, allowing users to observe any similarities and dissimilarities. Below, users will see a list of cancer types that contain the best-matched signature.
We use iMutSig to identify the most similar signature for a given PM/COSMIC signature or a user-supplied signature. Figure 2 shows the input panel after inputting COSMIC v3 signature SBS1 and Figure 3 shows the input panel after inputting PM signature P1. If users provide a user-supplied signature of either COSMIC-kind or PM-kind, the results can be seen in Figure 4 and Figure 5. Consider the example shown in Figure 4, where we input COSMIC v2 signature C1. iMutSig returned the most similar signatures COSMIC v2 signature C1, COSMIC v3 signature SBS1, and PM signature P7 (similarity = 1.000, 0.947, and 0.948, respectively) along with the names of its associated cancer types. When providing PM signature P1, iMutSig returned COSMIC v2 signature C10, v3 signature C10a and PM signature P1 (similarity = 0.816, 0.957, 1.0, respectively).
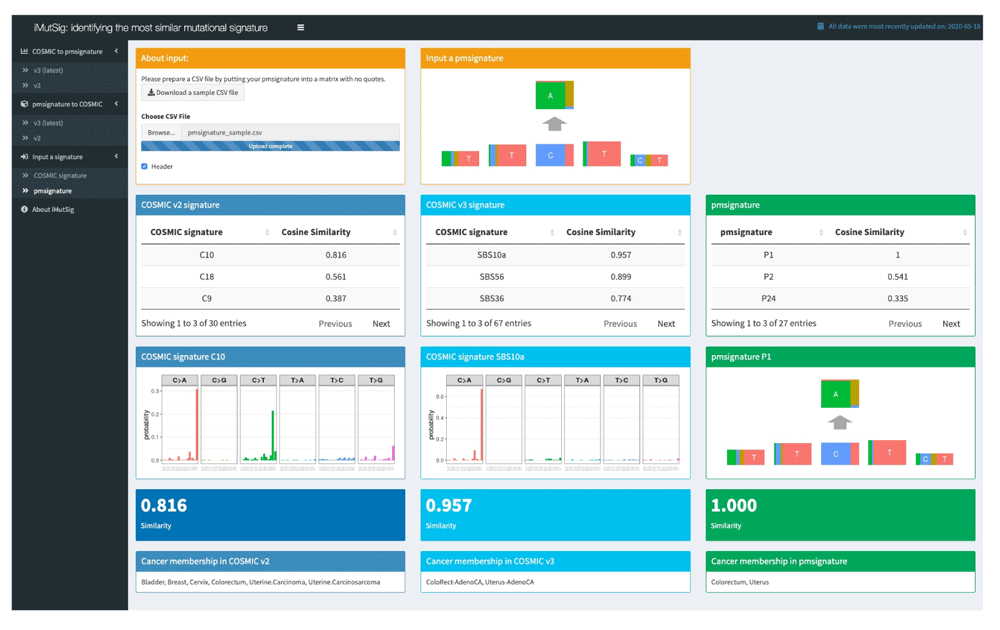
iMutSig is a user-friendly interactive browser-based application that allows users who have a signature that they have discovered in an analysis of their own data to identify the best-matched existing mutational signature from the COSMIC and PM databases. It also allows users to directly compare signatures between the two databases. It does this in an interactive way, and also allows straightforward visualization of results. iMutSig enables researchers to easily identify the most similar mutational signature and to easily access characteristic information from both data sources without additional software installation and programming of their own.
All data underlying the results are available as part of the article and no additional source data are required.
Software available from: https://zhiyang.shinyapps.io/iMutSig/
Source code available from: http://www.github.com/USCbiostats/iMutSig
Archived source code at time of publication: https://doi.org/10.5281/zenodo.387388816
License: MIT
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
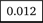
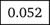
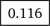
Comments on this article Comments (0)