Pomytkin I and Pinelis V. Insulin Receptors and Intracellular Ca2+ Form a Double-Negative Regulatory Feedback Loop Controlling Insulin Sensitivity [version 2; peer review: 3 approved]. F1000Research 2021, 9:598 (https://doi.org/10.12688/f1000research.24558.2)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
1Department of Advanced Cell Technologies, Sechenov First Moscow State Medical University (Sechenov University), Moscow, Moscow, 119991, Russian Federation 2National Medical Research Center for Children’s Health, Russian Ministry of Health, Moscow, 119991, Russian Federation
Igor Pomytkin
Roles:
Conceptualization,
Writing – Original Draft Preparation
Since the discovery of insulin and insulin receptors (IR) in the brain in 1978, numerous studies have revealed a fundamental role of IR in the central nervous system and its implication in regulating synaptic plasticity, long-term potentiation and depression, neuroprotection, learning and memory, and energy balance. Central insulin resistance has been found in diverse brain disorders including Alzheimer’s disease (AD). Impaired insulin signaling in AD is evident in the activation states of IR and downstream signaling molecules. This is mediated by Aβ oligomer-evoked Ca2+ influx by activating N-methyl-D-aspartate receptors (NMDARs) with Aβ oligomers directly, or indirectly through Aβ-induced release of glutamate, an endogenous NMDAR ligand. In the present opinion article, we highlight evidence that IR activity and free intracellular Ca2+ concentration [Ca2+]i form a double-negative regulatory feedback loop controlling insulin sensitivity, in which mitochondria play a key role, being involved in adenosine triphosphate (ATP) synthesis and IR activation. We found recently that the glutamate-evoked rise in [Ca2+]i inhibits activation of IR and, vice versa, insulin-induced activation of IR inhibits the glutamate-evoked rise in [Ca2+]i. In theory, such a double-negative regulatory feedback loop predicts that any condition leading to an increase of [Ca2+]i may trigger central insulin resistance and explains why central insulin resistance is implicated in the pathogenesis of AD, with which glutamate excitotoxicity is a comorbid condition. This model also predicts that any intervention aiming to maintain low [Ca2+]i may be useful for treating central insulin resistance.
The manuscript has been revised in accordance with reviewer's notes that (a) bistability is not a necessary consecuence of a double negative regulatory feedback loop and (b) figure 1 will be more useful when signal transduction pathways will be shown. Sentences related to "bistability" have been removed from the abstract, main text, and reference list. Figure 1 has been revised and includes now pathways described in the text of the first version.
The manuscript has been revised in accordance with reviewer's notes that (a) bistability is not a necessary consecuence of a double negative regulatory feedback loop and (b) figure 1 will be more useful when signal transduction pathways will be shown. Sentences related to "bistability" have been removed from the abstract, main text, and reference list. Figure 1 has been revised and includes now pathways described in the text of the first version.
Since the discovery of insulin1 and insulin receptors (IR)2 in the brain in 1978, numerous studies have revealed a fundamental role of IR in the central nervous system (CNS). IR-mediated signaling is implicated in the regulation of diverse functions in the CNS, including synaptic plasticity, long-term potentiation and depression, neuroprotection, learning and memory, and energy balance3. Central insulin resistance has been found in neurodegenerative diseases such as Alzheimer’s disease (AD)4,5 and Parkinson’s disease (PD)6, stroke, and traumatic brain injury (TBI)7. Impaired insulin signaling in AD is evident in the activation states of IR and downstream signaling molecules5. Compared with control cases, insulin in AD brains induced 24–58% less activation at the level of IR and 90% less activation of insulin receptor substrate 1 (IRS-1)5. It has been presumed5 that the inhibition of IR activation is mediated by Aβ oligomer-triggered Ca2+ influx, in part by activating N-methyl-D-aspartate receptors (NMDARs)8, followed by a rise in Akt1 pS4739, which can inhibit insulin-induced IR activation through Thr phosphorylation of the IR β subunit10. Aβ oligomers may activate the NMDAR-gated Ca2+ influx directly11 or indirectly through the intermediate release of glutamate, a ligand of NMDAR11–15. This suggests that the rise in intracellular free Ca2+ concentration ([Ca2+]i), evoked by either Aβ oligomers or glutamate, leads to dysfunctional activation of IR in AD. In the present opinion article, we highlight evidence that IR and [Ca2+]i form a double-negative regulatory feedback loop controlling insulin sensitivity, and mitochondria have a key role in this feedback loop, being involved in adenosine triphosphate (ATP) synthesis and IR activation.
Glutamate-evoked rise in [Ca2+]i causes inhibition of IR activation
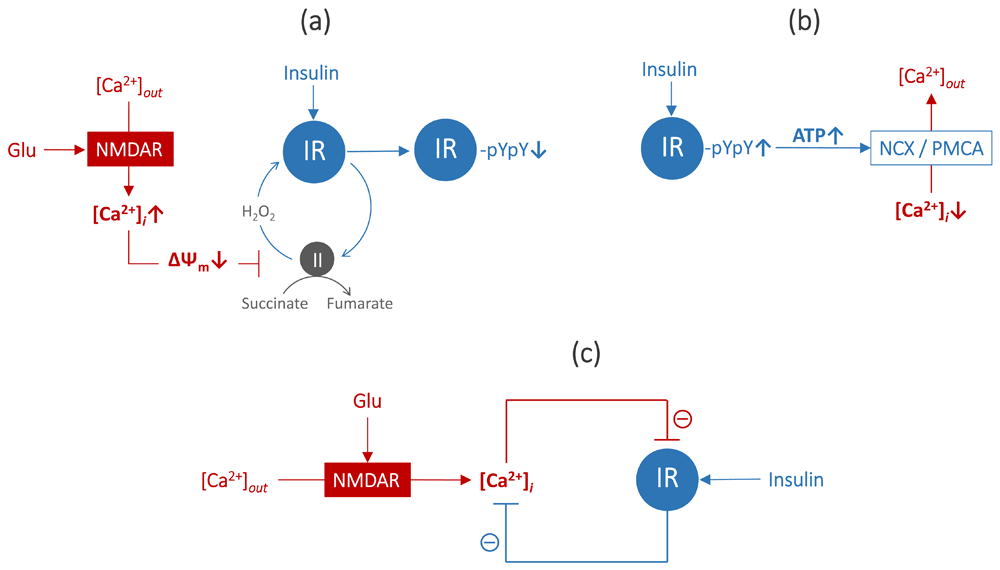
Glutamate serves as the major excitatory neurotransmitter in the CNS. Its excessive accumulation in a synaptic cleft can trigger excitotoxicity, a pathologic process leading to neuronal cell death. Glutamate-induced activation of the NMDAR-gated Ca2+ influx is generally considered central to the development of excitotoxicity16. Prolonged glutamate exposure causes a rapid initial increase in the [Ca2+]i, followed by a larger secondary [Ca2+]i increase concomitant with a decrease in the mitochondrial inner membrane potential (ΔΨm)17–19. We recently found that on Ca2+-induced mitochondrial depolarization, insulin induced 48% less activation of IR (assessed by pY1150/1151) compared with control20. Earlier, we showed that a decrease in ΔΨm can abrogate IR activation18, since the ΔΨm-dependent hydrogen peroxide (H2O2) mitochondrial signal at complex II is critically involved in the activation of IR in neurons21–23. Thus, the glutamate-evoked increase in [Ca2+]i, followed by the drop in ΔΨm, leads to the inhibition of insulin-induced activation of IR (Figure 1a).
Insulin prevents glutamate-evoked rise in [Ca2+]i
Normally, the NMDAR-gated Ca2+ influx is counterbalanced with Ca2+ efflux, which is governed by plasma membrane Ca2+ ATPase (PMCA) and the Na+/Ca2+ exchanger (NCX)24,25. NCX-mediated Ca2+ efflux is also ATP-dependent, since NCX exchanges one Ca2+ for three Na+, and the three Na+ are then pumped out by the Na+/K+ ATPase at the expense of one ATP. In excitotoxicity, prolonged stimulation with glutamate leads to ATP depletion and an abnormal rise in [Ca2+]i, since the massive Ca2+ influx is no longer counterbalanced by Ca2+ efflux26. Therefore, maintenance of ATP production is crucial for preventing the rise in [Ca2+]i in excitotoxicity. We found recently that pre-treatment with insulin prevents neurons from glutamate-evoked ATP depletion due to its protective effect on spare respiratory capacity (SRC), a measure that relates to the amount of extra ATP that can be produced via oxidative phosphorylation in case of increased energy demand19. The effect of insulin on SRC relates to its action on mitochondrial metabolism. It has long been known that the tricarboxylic acid cycle is the intracellular site of insulin action and that insulin acutely stimulates succinate oxidation at mitochondrial complex II26,27. Succinate oxidation at mitochondrial complex II has been identified recently as the main source of SRC28. In line with this, insulin prevented the glutamate-evoked rise in [Ca2+]i in our experiments with glutamate excitotoxicity19 (Figure 1b).
IR and [Ca2+]i form a double-negative feedback loop controlling insulin sensitivity
Collectively, this evidence suggests that a double-negative regulatory feedback loop exists between IR activity and [Ca2+]i. The glutamate-evoked rise in [Ca2+]i inhibits activation of IR and, vice versa, insulin-induced activation of IR inhibits the glutamate-evoked rise in [Ca2+]i (Figure 1c).
Figure 1. A double-negative feedback loop between insulin receptors (IR) activity and intracellular free Ca2+ concentration [Ca2+]i.
(a) glutamate-evoked increase in [Ca2+]i, followed by drop in ΔΨm, leads to inhibition of insulin-induced H2O2 signal at mitochondrial complex II, thereby inhibiting IR activation (IR-pYpY↓)20,21; (b) insulin-triggered increase in ATP levels enhances ATP-dependent Ca2+ efflux via PMCA and NCX, thereby lowering [Ca2+]i19; (c) glutamate triggers NMDA receptor-gated Ca2+ influx, inhibiting IR activation, and insulin triggers activation of IR, inhibiting [Ca2+]i rise. Abbreviations: ΔΨm , mitochondrial inner membrane potential; H2O2, hydrogen peroxide; pY, phosphotyrosine; ATP, adenosine triphosphate; PMCA, plasma membrane Ca2+ ATPase; NCX, Na +/Ca2+ exchanger; NMDA, N-methyl-D-aspartate.
This double-negative feedback loop model predicts that any condition leading to an increase in [Ca2+]i may trigger insulin resistance. It appears to explain why central insulin resistance is implicated in the pathogenesis of disorders such as AD4,5, PD6, stroke, and TBI7, with which glutamate excitotoxicity is a comorbid condition29. The model also predicts that any intervention aiming to prevent Ca2+ influx of or enhance efflux of Ca2+ from neurons, thereby maintaining low [Ca2+]i, may be useful for treating central insulin resistance. Given that Ca2+ efflux is ATP-dependent, any intervention directed to enhance ATP production in neurons may be especially useful to improve insulin sensitivity in the brain.
2.
Havrankova J, Roth J, Brownstein M:
Insulin receptors are widely distributed in the central nervous system of the rat.
Nature.
1978; 272(5656): 827–829. PubMed Abstract
| Publisher Full Text
3.
Pomytkin I, Costa-Nunes JP, Kasatkin V, et al.:
Insulin receptor in the brain: Mechanisms of activation and the role in the CNS pathology and treatment.
CNS Neurosci Ther.
2018; 24(9): 763–774. PubMed Abstract
| Publisher Full Text
| Free Full Text
4.
Arnold SE, Arvanitakis Z, Macauley-Rambach SL, et al.:
Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums.
Nat Rev Neurol.
2018; 14(3): 168–181. PubMed Abstract
| Publisher Full Text
| Free Full Text
5.
Talbot K, Wang HY, Kazi H, et al.:
Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline.
J Clin Invest.
2012; 122(4): 1316–38. PubMed Abstract
| Publisher Full Text
| Free Full Text
6.
Athauda D, Foltynie T:
Insulin resistance and Parkinson's disease: A new target for disease modification?
Prog Neurobiol.
2016; 145–146: 98–120. PubMed Abstract
| Publisher Full Text
8.
Alberdi E, Sánchez-Gómez MV, Cavaliere F, et al.:
Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors.
Cell Calcium.
2010; 47(3): 264–72. PubMed Abstract
| Publisher Full Text
9.
Perkinton MS, Ip JK, Wood GL, et al.:
Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones.
J Neurochem.
2002; 80(2): 239–54. PubMed Abstract
| Publisher Full Text
10.
Morisco C, Condorelli G, Trimarco V, et al.:
Akt mediates the cross-talk between beta-adrenergic and insulin receptors in neonatal cardiomyocytes.
Circ Res.
2005; 96(2): 180–8. PubMed Abstract
| Publisher Full Text
11.
Texidó L, Martín-Satué M, Alberdi E, et al.:
Amyloid β peptide oligomers directly activate NMDA receptors.
Cell Calcium.
2011; 49(3): 184–90. PubMed Abstract
| Publisher Full Text
12.
Zhao WQ, De Felice FG, Fernandez S, et al.:
Amyloid beta oligomers induce impairment of neuronal insulin receptors.
FASEB J.
2008; 22(1): 246–60. PubMed Abstract
| Publisher Full Text
13.
Kabogo D, Rauw G, Amritraj A, et al.:
ß-amyloid-related peptides potentiate K+-evoked glutamate release from adult rat hippocampal slices.
Neurobiol Aging.
2010; 31(7): 1164–72. PubMed Abstract
| Publisher Full Text
14.
Findley CA, Bartke A, Hascup KN, et al.:
Amyloid Beta-Related Alterations to Glutamate Signaling Dynamics During Alzheimer's Disease Progression.
ASN Neuro.
2019; 11: 1759091419855541. PubMed Abstract
| Publisher Full Text
| Free Full Text
15.
Hascup KN, Hascup ER:
Soluble Amyloid-β42 Stimulates Glutamate Release through Activation of the α7 Nicotinic Acetylcholine Receptor.
J Alzheimers Dis.
2016; 53(1): 337–47. PubMed Abstract
| Publisher Full Text
16.
Nicholls DG:
Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures.
Curr Mol Med.
2004; 4(2): 149–77. PubMed Abstract
| Publisher Full Text
17.
Vergun O, Keelan J, Khodorov BI, et al.:
Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones.
J Physiol.
1999; 519(Pt 2): 451–66. PubMed Abstract
| Publisher Full Text
| Free Full Text
18.
Abramov AY, Duchen MR:
Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity.
Biochim Biophys Acta.
2008; 1777(7–8): 953–64. PubMed Abstract
| Publisher Full Text
19.
Krasil'nikova I, Surin A, Sorokina E, et al.:
Insulin Protects Cortical Neurons Against Glutamate Excitotoxicity.
Front Neurosci.
2019; 13: 1027. PubMed Abstract
| Publisher Full Text
| Free Full Text
20.
Pomytkin I, Krasil'nikova I, Bakaeva Z, et al.:
Excitotoxic glutamate causes neuronal insulin resistance by inhibiting insulin receptor/Akt/mTOR pathway.
Mol Brain.
2019; 12(1): 112. PubMed Abstract
| Publisher Full Text
| Free Full Text
21.
Persiyantseva NA, Storozhevykh TP, Senilova YE, et al.:
Mitochondrial H2O2 as an enable signal for triggering autophosphorylation of insulin receptor in neurons.
J Mol Signal.
2013; 8(1): 11. PubMed Abstract
| Publisher Full Text
| Free Full Text
22.
Storozhevykh TP, Senilova YE, Persiyantseva NA, et al.:
Mitochondrial respiratory chain is involved in insulin-stimulated hydrogen peroxide production and plays an integral role in insulin receptor autophosphorylation in neurons.
BMC Neurosci.
2007; 8: 84. PubMed Abstract
| Publisher Full Text
| Free Full Text
24.
Carafoli E, Santella L, Branca D, et al.:
Generation, control, and processing of cellular calcium signals.
Crit Rev Biochem Mol Biol.
2001; 36(2): 107–260. PubMed Abstract
| Publisher Full Text
25.
Brittain MK, Brustovetsky T, Sheets PL, et al.:
Delayed calcium dysregulation in neurons requires both the NMDA receptor and the reverse Na+/Ca2+ exchanger.
Neurobiol Dis.
2012; 46(1): 109–17. PubMed Abstract
| Publisher Full Text
| Free Full Text
26.
Bessman SP, Mohan C, Zaidise I:
Intracellular site of insulin action: mitochondrial Krebs cycle.
Proc Natl Acad Sci U S A.
1986; 83(14): 5067–70. PubMed Abstract
| Publisher Full Text
| Free Full Text
27.
Bessman SP, Mohan C:
Insulin as a probe of mitochondrial metabolism in situ.
Mol Cell Biochem.
1997; 174(1–2): 91–6. PubMed Abstract
| Publisher Full Text
28.
Pfleger J, He M, Abdellatif M:
Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival.
Cell Death Dis.
2015; 6(7): e1835. PubMed Abstract
| Publisher Full Text
| Free Full Text
1
Department of Advanced Cell Technologies, Sechenov First Moscow State Medical University (Sechenov University), Moscow, Moscow, 119991, Russian Federation 2
National Medical Research Center for Children’s Health, Russian Ministry of Health, Moscow, 119991, Russian Federation
Igor Pomytkin
Roles:
Conceptualization,
Writing – Original Draft Preparation
Pomytkin I and Pinelis V. Insulin Receptors and Intracellular Ca2+ Form a Double-Negative Regulatory Feedback Loop Controlling Insulin Sensitivity [version 2; peer review: 3 approved]. F1000Research 2021, 9:598 (https://doi.org/10.12688/f1000research.24558.2)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
Although the authors have adequately presented point-by-point responses to my initial concerns, they have taken a minimalistic approach to incorporating some of these concepts into the actual manuscript. It would have been interesting to read the authors ideas on how
... Continue reading
Although the authors have adequately presented point-by-point responses to my initial concerns, they have taken a minimalistic approach to incorporating some of these concepts into the actual manuscript. It would have been interesting to read the authors ideas on how this double feedback loop is challenged in different disease states considering this forms the premise of the introduction and the reasoning behind the opinion piece. Regardless, in situations of extreme deviations from homeostatic calcium concentrations (stroke and traumatic brain injury) concerns about cerebral insulin resistance is minimal compared to the rapid loss of nervous tissue. This double feedback loop postulate may be better suited for slower progressing neurodegenerative disorders.
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
The manuscript can now be accepted
... Continue reading
The manuscript can now be accepted as all concerns have been addressed.
Competing Interests: No competing interests were disclosed.
Reviewer Expertise: Systems biology, Liver metabolism, modeling signaling pathways
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
The authors propose an existence of double negative feedback loop which may result in bistable response. It is clear that Ca signaling and insulin signaling play a role in the CNS and the related pathogenesis of neurogenerative diseases. The mitochondrial
... Continue reading
The authors propose an existence of double negative feedback loop which may result in bistable response. It is clear that Ca signaling and insulin signaling play a role in the CNS and the related pathogenesis of neurogenerative diseases. The mitochondrial function and thereby ATP synthesis also plays a role in the stated phenotype. The proposed dual negative feedback on each other can toggle between two states, but it may not be bistable. Bistable is with respective to an input variation. It is unclear what is the input that the hypothesis is being discussed. At a given input, the existence of both steady state, with different history, yields bistability. A figure that demonstrates all the molecular components and action including mitochondrial state and metabolites is more helpful than what is illustrated.
Is the topic of the opinion article discussed accurately in the context of the current literature?
Yes
Are all factual statements correct and adequately supported by citations?
Yes
Are arguments sufficiently supported by evidence from the published literature?
Yes
Are the conclusions drawn balanced and justified on the basis of the presented arguments?
No
Competing Interests: No competing interests were disclosed.
Reviewer Expertise: Systems biology, Liver metabolism, modeling signaling pathways
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard, however I have significant reservations, as outlined above.
Igor Pomytkin, Department of Advanced Cell Technologies, Sechenov First Moscow State Medical University (Sechenov University), Moscow, 119991, Russian Federation
13 Jan 2021
Author Response
The manuscript has been revised in accordance with notes that (a) bistability is not a necessary consecuence of a double negative regulatory feedback loop and (b) figure 1 will be
...
Continue readingThe manuscript has been revised in accordance with notes that (a) bistability is not a necessary consecuence of a double negative regulatory feedback loop and (b) figure 1 will be more useful when signal transduction pathways will be shown. Sentences related to "bistability" have been removed from the abstract, main text, and reference list. Figure 1 has been revised and includes now pathways described in the text of the first version.
The manuscript has been revised in accordance with notes that (a) bistability is not a necessary consecuence of a double negative regulatory feedback loop and (b) figure 1 will be more useful when signal transduction pathways will be shown. Sentences related to "bistability" have been removed from the abstract, main text, and reference list. Figure 1 has been revised and includes now pathways described in the text of the first version.
Igor Pomytkin, Department of Advanced Cell Technologies, Sechenov First Moscow State Medical University (Sechenov University), Moscow, 119991, Russian Federation
13 Jan 2021
Author Response
The manuscript has been revised in accordance with notes that (a) bistability is not a necessary consecuence of a double negative regulatory feedback loop and (b) figure 1 will be
...
Continue readingThe manuscript has been revised in accordance with notes that (a) bistability is not a necessary consecuence of a double negative regulatory feedback loop and (b) figure 1 will be more useful when signal transduction pathways will be shown. Sentences related to "bistability" have been removed from the abstract, main text, and reference list. Figure 1 has been revised and includes now pathways described in the text of the first version.
The manuscript has been revised in accordance with notes that (a) bistability is not a necessary consecuence of a double negative regulatory feedback loop and (b) figure 1 will be more useful when signal transduction pathways will be shown. Sentences related to "bistability" have been removed from the abstract, main text, and reference list. Figure 1 has been revised and includes now pathways described in the text of the first version.
This model predicts that any disease that causes elevated [Ca2+]i may trigger central insulin resistance, and explains why central insulin resistance is related to the pathogenesis of AD, and glutamate excitotoxicity is a comorbidity. The model also predicts that any
... Continue reading
This model predicts that any disease that causes elevated [Ca2+]i may trigger central insulin resistance, and explains why central insulin resistance is related to the pathogenesis of AD, and glutamate excitotoxicity is a comorbidity. The model also predicts that any intervention aimed at maintaining low [Ca2+]i can be used to treat central insulin resistance.
It is an interesting theory between brain insulin resistance and AD. Although not much experimental data support the theory directly, it is worth following.
Is the topic of the opinion article discussed accurately in the context of the current literature?
Yes
Are all factual statements correct and adequately supported by citations?
Yes
Are arguments sufficiently supported by evidence from the published literature?
Yes
Are the conclusions drawn balanced and justified on the basis of the presented arguments?
Yes
Competing Interests: No competing interests were disclosed.
Reviewer Expertise: cerebral vascular disease
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
The authors put forth an opinion article in regard to a double negative feedback loop that controls cerebral insulin receptor signaling during times of either high glutamate or insulin levels. They briefly go onto hypothesize that any disease state resulting
... Continue reading
The authors put forth an opinion article in regard to a double negative feedback loop that controls cerebral insulin receptor signaling during times of either high glutamate or insulin levels. They briefly go onto hypothesize that any disease state resulting in elevated intracellular calcium will result in reduced cerebral insulin resistance, particularly when excitotoxicity is a comorbid factor. The authors attempt to simplify an extremely complicated phenomenon. However, this oversimplification neglects to take into account several factors that need to be addressed.
Reduced insulin signaling as a result for glutamate induced calcium influx has been discussed in other literature (see Zhao et al., 20081). This work should be taken into consideration when authors discuss their model.
Although insulin receptors are ubiquitously expressed in different cell types of the CNS, these cell types rely on different mechanisms for ATP production. Glial cells predominantly use glycolytic pathways in the cytoplasm whereas neurons rely on oxidative phosphorylation in the mitochondria. (For review see Pellerin and Magistretti, 20122). Accordingly, the cellular localization of ATP production should be taken into consideration. Furthermore, do the authors believe different cell type specific mechanisms exist that their model should take into consideration?
In several places, the authors discuss the role of amyloid on activation of NMDA receptors and the subsequent intracellular Calcium increase. However, the authors have not taken into account how amyloid binding can also elicit glutamate release from either α7nAChRs (see Hascup and Hascup, 20163 and Mura et al., 20124) or mGLUR5 receptors (Renner et al., 20105). In the latter case, several laboratories have shown mGLUR5 acts as a scaffolding complex for amyloid accumulation causing receptor clustering at the membrane surface and results in elevated intracellular calcium levels.
Additionally, the manuscript could be improved if a discussion on how their model might vary across different CNS disorders such as AD, Parkinson’s TBI, etc. in relation to healthy functional activity.
The authors neglect to take into account factors that also play a role in modulating insulin signaling. These include:
Inflammation. This is prominent in numerous disease states and can negatively impact insulin signaling, while exacerbating mechanisms associated with multiple neurodegenerative disorders.
Inhibitory feedback regulation. Insulin signaling is tightly controlled to prevent perturbations in metabolism as well as control the specificity of the signal on multiple downstream effectors. Several phosphatases are responsible for this, not just at the receptor, but also on effector enzymes.The current model does not take into consideration this tightly controlled feedback loop.
Receptor internalization. Upon insulin binding, the insulin receptor becomes internalized as another means to control the strength and duration of the signal. This internalization is more prominent in hyperinsulinemia and may account for the resulting insulin resistance. How would this model change during stages of insulin resistance, which are hypothesized to initiate the cognitive decline observed in AD?
Peripheral insulin signaling.Insulin produced in the pancreas is able to enter the CNS.How does the proposed model take into consideration fluctuations during normal periods of food consumption?
Are the authors proposing a similar mechanism for the structurally analogous insulin-like growth factor-1?
Minor concerns:
The glutamate pathway in Figure 1 should have a different color scheme to make it easier to differentiate from Calcium concentration.
Figure 1B is slightly confusing. The red line makes it seem that low levels of glutamate give rise to high levels of Calcium instead of showing just the rise in calcium. A way to incorporate glutamate activation of NMDA receptors in Fig 1B & C might help to conceptualize the model.
The abstract is lengthy in relation to the article. I would suggest this is shortened and made more succinct.
Is the topic of the opinion article discussed accurately in the context of the current literature?
Partly
Are all factual statements correct and adequately supported by citations?
Yes
Are arguments sufficiently supported by evidence from the published literature?
No
Are the conclusions drawn balanced and justified on the basis of the presented arguments?
Partly
References
1. Zhao WQ, De Felice FG, Fernandez S, Chen H, et al.: Amyloid beta oligomers induce impairment of neuronal insulin receptors.FASEB J. 2008; 22 (1): 246-60 PubMed Abstract | Publisher Full Text 2. Pellerin L, Magistretti PJ: Sweet sixteen for ANLS.J Cereb Blood Flow Metab. 2012; 32 (7): 1152-66 PubMed Abstract | Publisher Full Text 3. Hascup K, Hascup E: Soluble Amyloid-β42 Stimulates Glutamate Release through Activation of the α7 Nicotinic Acetylcholine Receptor. Journal of Alzheimer's Disease. 2016; 53 (1): 337-347 Publisher Full Text 4. Mura E, Zappettini S, Preda S, Biundo F, et al.: Dual effect of beta-amyloid on α7 and α4β2 nicotinic receptors controlling the release of glutamate, aspartate and GABA in rat hippocampus.PLoS One. 2012; 7 (1): e29661 PubMed Abstract | Publisher Full Text 5. Renner M, Lacor PN, Velasco PT, Xu J, et al.: Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5.Neuron. 2010; 66 (5): 739-54 PubMed Abstract | Publisher Full Text
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to state that I do not consider it to be of an acceptable scientific standard, for reasons outlined above.
Igor Pomytkin, Department of Advanced Cell Technologies, Sechenov First Moscow State Medical University (Sechenov University), Moscow, 119991, Russian Federation
10 Jul 2020
Author Response
Our opinion article relates only to relationship between intracellular Ca2+ concentrations [Ca2+]i and insulin receptor activation state, and not between [Ca2+]i and insulin signaling system as a whole. Insulin resistance
...
Continue readingOur opinion article relates only to relationship between intracellular Ca2+ concentrations [Ca2+]i and insulin receptor activation state, and not between [Ca2+]i and insulin signaling system as a whole. Insulin resistance is the term that currently applied to any of the biological actions of insulin and, therefore, is too broad to be discussed in terms of models that may predict the system behavior. The proposed model in the opinion article is not oversimplified, but takes into consideration only link between [Ca2+]i and the stage of activation of insulin receptor kinase (i.e Tyr1150/1151 phosphorylation), the earliest step in insulin action that precedes all other signaling events and effects of insulin.
In our opinion article we selected only two measurable parameters, namely [Ca2+]i and insulin receptor activation state defined as Tyr1150/1151 phosphorylation, but not downstream molecules of IR signaling pathway such as IRS-1 or others. Therefore, our opinion relates only to the activation of insulin receptor, and not to downstream molecules or events. This approach relates directly to insulin sensitivity, since the activation of the receptor with insulin is the only stage at which insulin sensitivity can be measured directly.
The opinion about existence of regulatory double-negative feedback loop between [Ca2+]i and insulin receptor activation state is based on our experimental results, obtained at the same experimental conditions in two studies that have already been published [references 19 and 20].
Results of Zhao et al. do not contradict our opinion about relationship between Ca2+ and insulin receptor activation state. Moreover, results of Zhao et al. support our opinion. Zhao et al. have found that glutamate stimulation reduced the insulin-stimulated tyrosine phosphorylation of the receptor beta-subunit at Tyr1150/1151 and this glutamate effect was completely inhibited by the cell-permeable Ca2+ chelator BAPTA-AM. Although authors did not measure intracellular Ca2+ concentrations, they concluded that IR inhibition is dependent on elevated intracellular Ca2+, given a well-known link between glutamate and Ca2+. It is the same conclusion that we made. The difference is only that our conclusion is made on our direct measurements of intracellular Ca2+.
In our studies [references 19 and 20] that underlie the our opinion we used glia-free cortical neurons and directly measured ATP levels.
Our opinion relates to link between [Ca2+]i and insulin receptor activation state, and amyloid-NMDA relationship are out of scope of our opinion.
Our opinion is limited to only insulin receptor activation, but not to more broad “insulin signaling”. According to current knowledge, inflammation affects insulin signaling, but downstream of insulin receptor at IRS-1 level. This is out of scope of our opinion.
Our opinion does not relate to any signaling events downstream of insulin receptor, such phosphatase action and receptor internalization.
The insulin regulated internalization of insulin receptors has been shown to require autophosphorylation of all three regulatory tyrosines 1146, 1150, and 1151. Carpentier JL et al. Two steps of insulin receptor internalization depend on different domains of the beta-subunit. J Cell Biol. 1993 Sep;122(6):1243-52. doi: 10.1083/jcb.122.6.1243. Thus, the internalization occurs after the activation of IR and, therefore, is out of the scope of our opinion.
According to the model, insulin inhibits rise of [Ca2+]i independently of the insulin source. Therefore, during periods of insulin transport to the brain, the rise of [Ca2+]i , e.g. glutamate-evoked, would be diminished, independently on whether it comes from pancreas or administered intranasally.
We did not propose the same mechanism for IGF-1 in our opinion aticle, since we have no supportive evidence. However, it is likely that there is a link between [Ca2+]i and activation state of IGFR1.
In a conclusion, the model proposed in the opinion article relates only to relationship beteen two measurable parameters, namely intracellular Ca2+ concentrations and insulin receptor activation state. Therefore, all other downstream elements of insulin signaling system and factors affecting the downstream elements are out of scope of this opinion. The opinion is completely based on our experimental results previously published, references 19 and 20 of the opinion article.
Our opinion article relates only to relationship between intracellular Ca2+ concentrations [Ca2+]i and insulin receptor activation state, and not between [Ca2+]i and insulin signaling system as a whole. Insulin resistance is the term that currently applied to any of the biological actions of insulin and, therefore, is too broad to be discussed in terms of models that may predict the system behavior. The proposed model in the opinion article is not oversimplified, but takes into consideration only link between [Ca2+]i and the stage of activation of insulin receptor kinase (i.e Tyr1150/1151 phosphorylation), the earliest step in insulin action that precedes all other signaling events and effects of insulin.
In our opinion article we selected only two measurable parameters, namely [Ca2+]i and insulin receptor activation state defined as Tyr1150/1151 phosphorylation, but not downstream molecules of IR signaling pathway such as IRS-1 or others. Therefore, our opinion relates only to the activation of insulin receptor, and not to downstream molecules or events. This approach relates directly to insulin sensitivity, since the activation of the receptor with insulin is the only stage at which insulin sensitivity can be measured directly.
The opinion about existence of regulatory double-negative feedback loop between [Ca2+]i and insulin receptor activation state is based on our experimental results, obtained at the same experimental conditions in two studies that have already been published [references 19 and 20].
Results of Zhao et al. do not contradict our opinion about relationship between Ca2+ and insulin receptor activation state. Moreover, results of Zhao et al. support our opinion. Zhao et al. have found that glutamate stimulation reduced the insulin-stimulated tyrosine phosphorylation of the receptor beta-subunit at Tyr1150/1151 and this glutamate effect was completely inhibited by the cell-permeable Ca2+ chelator BAPTA-AM. Although authors did not measure intracellular Ca2+ concentrations, they concluded that IR inhibition is dependent on elevated intracellular Ca2+, given a well-known link between glutamate and Ca2+. It is the same conclusion that we made. The difference is only that our conclusion is made on our direct measurements of intracellular Ca2+.
In our studies [references 19 and 20] that underlie the our opinion we used glia-free cortical neurons and directly measured ATP levels.
Our opinion relates to link between [Ca2+]i and insulin receptor activation state, and amyloid-NMDA relationship are out of scope of our opinion.
Our opinion is limited to only insulin receptor activation, but not to more broad “insulin signaling”. According to current knowledge, inflammation affects insulin signaling, but downstream of insulin receptor at IRS-1 level. This is out of scope of our opinion.
Our opinion does not relate to any signaling events downstream of insulin receptor, such phosphatase action and receptor internalization.
The insulin regulated internalization of insulin receptors has been shown to require autophosphorylation of all three regulatory tyrosines 1146, 1150, and 1151. Carpentier JL et al. Two steps of insulin receptor internalization depend on different domains of the beta-subunit. J Cell Biol. 1993 Sep;122(6):1243-52. doi: 10.1083/jcb.122.6.1243. Thus, the internalization occurs after the activation of IR and, therefore, is out of the scope of our opinion.
According to the model, insulin inhibits rise of [Ca2+]i independently of the insulin source. Therefore, during periods of insulin transport to the brain, the rise of [Ca2+]i , e.g. glutamate-evoked, would be diminished, independently on whether it comes from pancreas or administered intranasally.
We did not propose the same mechanism for IGF-1 in our opinion aticle, since we have no supportive evidence. However, it is likely that there is a link between [Ca2+]i and activation state of IGFR1.
In a conclusion, the model proposed in the opinion article relates only to relationship beteen two measurable parameters, namely intracellular Ca2+ concentrations and insulin receptor activation state. Therefore, all other downstream elements of insulin signaling system and factors affecting the downstream elements are out of scope of this opinion. The opinion is completely based on our experimental results previously published, references 19 and 20 of the opinion article.
Competing Interests:there is no competing interestsClose
Igor Pomytkin, Department of Advanced Cell Technologies, Sechenov First Moscow State Medical University (Sechenov University), Moscow, 119991, Russian Federation
10 Jul 2020
Author Response
Our opinion article relates only to relationship between intracellular Ca2+ concentrations [Ca2+]i and insulin receptor activation state, and not between [Ca2+]i and insulin signaling system as a whole. Insulin resistance
...
Continue readingOur opinion article relates only to relationship between intracellular Ca2+ concentrations [Ca2+]i and insulin receptor activation state, and not between [Ca2+]i and insulin signaling system as a whole. Insulin resistance is the term that currently applied to any of the biological actions of insulin and, therefore, is too broad to be discussed in terms of models that may predict the system behavior. The proposed model in the opinion article is not oversimplified, but takes into consideration only link between [Ca2+]i and the stage of activation of insulin receptor kinase (i.e Tyr1150/1151 phosphorylation), the earliest step in insulin action that precedes all other signaling events and effects of insulin.
In our opinion article we selected only two measurable parameters, namely [Ca2+]i and insulin receptor activation state defined as Tyr1150/1151 phosphorylation, but not downstream molecules of IR signaling pathway such as IRS-1 or others. Therefore, our opinion relates only to the activation of insulin receptor, and not to downstream molecules or events. This approach relates directly to insulin sensitivity, since the activation of the receptor with insulin is the only stage at which insulin sensitivity can be measured directly.
The opinion about existence of regulatory double-negative feedback loop between [Ca2+]i and insulin receptor activation state is based on our experimental results, obtained at the same experimental conditions in two studies that have already been published [references 19 and 20].
Results of Zhao et al. do not contradict our opinion about relationship between Ca2+ and insulin receptor activation state. Moreover, results of Zhao et al. support our opinion. Zhao et al. have found that glutamate stimulation reduced the insulin-stimulated tyrosine phosphorylation of the receptor beta-subunit at Tyr1150/1151 and this glutamate effect was completely inhibited by the cell-permeable Ca2+ chelator BAPTA-AM. Although authors did not measure intracellular Ca2+ concentrations, they concluded that IR inhibition is dependent on elevated intracellular Ca2+, given a well-known link between glutamate and Ca2+. It is the same conclusion that we made. The difference is only that our conclusion is made on our direct measurements of intracellular Ca2+.
In our studies [references 19 and 20] that underlie the our opinion we used glia-free cortical neurons and directly measured ATP levels.
Our opinion relates to link between [Ca2+]i and insulin receptor activation state, and amyloid-NMDA relationship are out of scope of our opinion.
Our opinion is limited to only insulin receptor activation, but not to more broad “insulin signaling”. According to current knowledge, inflammation affects insulin signaling, but downstream of insulin receptor at IRS-1 level. This is out of scope of our opinion.
Our opinion does not relate to any signaling events downstream of insulin receptor, such phosphatase action and receptor internalization.
The insulin regulated internalization of insulin receptors has been shown to require autophosphorylation of all three regulatory tyrosines 1146, 1150, and 1151. Carpentier JL et al. Two steps of insulin receptor internalization depend on different domains of the beta-subunit. J Cell Biol. 1993 Sep;122(6):1243-52. doi: 10.1083/jcb.122.6.1243. Thus, the internalization occurs after the activation of IR and, therefore, is out of the scope of our opinion.
According to the model, insulin inhibits rise of [Ca2+]i independently of the insulin source. Therefore, during periods of insulin transport to the brain, the rise of [Ca2+]i , e.g. glutamate-evoked, would be diminished, independently on whether it comes from pancreas or administered intranasally.
We did not propose the same mechanism for IGF-1 in our opinion aticle, since we have no supportive evidence. However, it is likely that there is a link between [Ca2+]i and activation state of IGFR1.
In a conclusion, the model proposed in the opinion article relates only to relationship beteen two measurable parameters, namely intracellular Ca2+ concentrations and insulin receptor activation state. Therefore, all other downstream elements of insulin signaling system and factors affecting the downstream elements are out of scope of this opinion. The opinion is completely based on our experimental results previously published, references 19 and 20 of the opinion article.
Our opinion article relates only to relationship between intracellular Ca2+ concentrations [Ca2+]i and insulin receptor activation state, and not between [Ca2+]i and insulin signaling system as a whole. Insulin resistance is the term that currently applied to any of the biological actions of insulin and, therefore, is too broad to be discussed in terms of models that may predict the system behavior. The proposed model in the opinion article is not oversimplified, but takes into consideration only link between [Ca2+]i and the stage of activation of insulin receptor kinase (i.e Tyr1150/1151 phosphorylation), the earliest step in insulin action that precedes all other signaling events and effects of insulin.
In our opinion article we selected only two measurable parameters, namely [Ca2+]i and insulin receptor activation state defined as Tyr1150/1151 phosphorylation, but not downstream molecules of IR signaling pathway such as IRS-1 or others. Therefore, our opinion relates only to the activation of insulin receptor, and not to downstream molecules or events. This approach relates directly to insulin sensitivity, since the activation of the receptor with insulin is the only stage at which insulin sensitivity can be measured directly.
The opinion about existence of regulatory double-negative feedback loop between [Ca2+]i and insulin receptor activation state is based on our experimental results, obtained at the same experimental conditions in two studies that have already been published [references 19 and 20].
Results of Zhao et al. do not contradict our opinion about relationship between Ca2+ and insulin receptor activation state. Moreover, results of Zhao et al. support our opinion. Zhao et al. have found that glutamate stimulation reduced the insulin-stimulated tyrosine phosphorylation of the receptor beta-subunit at Tyr1150/1151 and this glutamate effect was completely inhibited by the cell-permeable Ca2+ chelator BAPTA-AM. Although authors did not measure intracellular Ca2+ concentrations, they concluded that IR inhibition is dependent on elevated intracellular Ca2+, given a well-known link between glutamate and Ca2+. It is the same conclusion that we made. The difference is only that our conclusion is made on our direct measurements of intracellular Ca2+.
In our studies [references 19 and 20] that underlie the our opinion we used glia-free cortical neurons and directly measured ATP levels.
Our opinion relates to link between [Ca2+]i and insulin receptor activation state, and amyloid-NMDA relationship are out of scope of our opinion.
Our opinion is limited to only insulin receptor activation, but not to more broad “insulin signaling”. According to current knowledge, inflammation affects insulin signaling, but downstream of insulin receptor at IRS-1 level. This is out of scope of our opinion.
Our opinion does not relate to any signaling events downstream of insulin receptor, such phosphatase action and receptor internalization.
The insulin regulated internalization of insulin receptors has been shown to require autophosphorylation of all three regulatory tyrosines 1146, 1150, and 1151. Carpentier JL et al. Two steps of insulin receptor internalization depend on different domains of the beta-subunit. J Cell Biol. 1993 Sep;122(6):1243-52. doi: 10.1083/jcb.122.6.1243. Thus, the internalization occurs after the activation of IR and, therefore, is out of the scope of our opinion.
According to the model, insulin inhibits rise of [Ca2+]i independently of the insulin source. Therefore, during periods of insulin transport to the brain, the rise of [Ca2+]i , e.g. glutamate-evoked, would be diminished, independently on whether it comes from pancreas or administered intranasally.
We did not propose the same mechanism for IGF-1 in our opinion aticle, since we have no supportive evidence. However, it is likely that there is a link between [Ca2+]i and activation state of IGFR1.
In a conclusion, the model proposed in the opinion article relates only to relationship beteen two measurable parameters, namely intracellular Ca2+ concentrations and insulin receptor activation state. Therefore, all other downstream elements of insulin signaling system and factors affecting the downstream elements are out of scope of this opinion. The opinion is completely based on our experimental results previously published, references 19 and 20 of the opinion article.
Competing Interests:there is no competing interestsClose
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)