Keywords
Acute myeloid leukemia, Triplex, DNA methylation, long non coding RNA, extra-coding CEBPα
This article is included in the Genomics and Genetics gateway.
This article is included in the Cell & Molecular Biology gateway.
Acute myeloid leukemia, Triplex, DNA methylation, long non coding RNA, extra-coding CEBPα
Figure 1b was updated. (Details: This was done to clarify the abundance of hypermethylated sites versus non-hypermethylated sites.)
Figure 1c was updated. (details: The x-axis was confusing and unclear, so we had to provide more explicit labels.)
A new image was added to figure 1 as figure 1d. (details: Using external data, we showed the relationship between ecCEBPA and CpG methylation on the predicted binding and non-binding sites.)
Since figure 1d was not present before now, the previous figures 1d and 1e were updated to 1E and 1F respectively.
Figure 2a was updated: (details: we provided annotations on the image to show the single-stranded region that was rich in triplex-forming oligonucleotides.)
A new image was added to figure 2 as Figure 2c. (Details: Through this, we showed the methylation difference in only the experimentally validated ecCEPBA contacts.)
Since figure 2c was not present before now, the previous figures 2c and 2d were updated to 2D and 2E respectively.
We also provided a table of some manually curated functional annotations for genes that are close to the experimentally validated CpGs
Discussion:
We discussed the new findings presented in figure 1d in the context of Di Ruscio's paper on ecCEBPA's expression and demethylation activities.
Underlying Data:
We provided the link to ENCODE data showing the normal methylome of HL-60 cells using RRBS.
See the authors' detailed response to the review by Oleg N. Demidov
See the authors' detailed response to the review by Amrita Singh
Acute myeloid leukemia (AML) is a malignant tumor characterized by the proliferation of undifferentiated myeloblasts1,2. It is the most prevalent form of leukemia in older adults (>60 years) with an annual mortality rate of 50% and a 5-year survival rate of 24%2,3. With the combined effects of the global increase in average life expectancy and AML drug inefficiency, the number of patients is expected to significantly increase in the coming years4,5.
The current understanding of the molecular interplay in AML has been defined under two distinct categories; (i) genetic abnormalities and (ii) non-random chromosomal rearrangements. Cases of AML with chromosomal rearrangements as t(15;17) [PML-RARA], t(9;22) [BCR-ABL], inv(16) [CBFB-MYH11], t(8;21) [RUNX1-ETO] are called cytogenetically abnormal (CA-AML), while cases with genetic abnormalities (including frequent mutations in DNMT3A, NPM1, CEBPα, IDH1/2, TET2, FLT3-ITD) are called cytogenetically normal (CN-AML)1,4. The former accounts for 50–55%, while the latter accounts for 45–50% of diagnosed AML cases6,7. Even though these mutations and chromosomal alterations are crucial for initiating AML, they are not sufficient to explain AML progression, heterogeneity, and relapse8.
Recently, studies have identified the role of non-coding RNAs, especially long non-coding RNAs (lncRNA) in the initiation and progression of cancers9–11. LncRNAs are emerging functional transcriptional products of at least 200 nucleotides lacking an open reading frame. Although they account for a large proportion of transcriptional products in mammals (about 58,000 loci)12, only a small number of lncRNAs have been well characterized. Although lncRNAs are mostly not conserved evolutionarily, they are heavily regulated suggesting their functional role12,13. They may function either as signal transducers, protein guides, or molecular scaffolds to regulate transcriptional and epigenetic events14–16. Some lncRNAs perform these functions in cis- by modulating transcription of nearby genes (Dum)17 or act in trans-, by modulating genes at multiple distant loci (MALAT1)18, while some can do both (HOTAIR)9,19.
Recent studies have identified lncRNAs for their remarkable role in regulating major epigenetic processes such as DNA methylation and chromatin remodeling. DNA methylation in mammals is coordinated by one of the three DNA methyl-transferases (DNMT); DNMT1, DNMT3A, and DNMT3B7,17,20. LncRNAs have been identified in recent studies as important agents that can modulate DNA methylation, either by activating or repressing DNMTs. For example, the lncRNA Dum was discovered to repress a nearby gene Dppa2 by recruiting multiple DNMTs leading to methylation of a promoter region, thus promoting myoblast differentiation17. Conversely, the lncRNA H19 represses the activity of DNMT3B by interacting with SAHH which hydrolyzes SAH, a step required for DNMT3B activation21. ecCEBPα, which is transcribed from the CEBPα locus, directly blocks DNMT1 to prevent methylation of proximal and distal located promoters, thus promoting CEBPα-mediated granulocyte differentiation20. Mechanistic studies via reduced representation bisulfite sequencing and RNA immunoprecipitation sequencing shows that ecCEBPα suppresses DNA methylation in cis- by acting as a shield that sequesters DNMT1 from the CEBPα promoter. We speculate that ecCEBPα could regulate DNMT1 activities in distant DNA regions (in trans-) as well. The mechanism of this potential interaction is to be determined.
In the recent version of GENCODE, ecCEBPα is not annotated most likely due to an overlap with a protein-coding gene CEBPα on the same strand. We retrieved the complete ecCEBPα sequence from the human genome (hg19, chr19: 33298573-33303358) based on information reported in the work of Di Ruscio et al.20. ecCEBPα is approximately 4.8kb and it overlaps with the intronless CEBPα gene (~2.6kb) on the same strand. ecCEBPα does not share either the same transcription start site (TSS) or transcription end site (TES) with CEBPα, starting ~0.89kb upstream and ending ~1.46kb downstream of the CEBPα gene.
CpG methylation (Illumina 450K array) and CEBPα mutation data for 186 AML patients were retrieved from the Cancer Genome Atlas (TCGA: http://firebrowse.org). CpG methylation levels were measured in 307796 unique loci. We split all the AML patients into two groups based on CEBPα mutation status (13 patients with a CEBPα mutation and 173 patients without a mutation). We classified a CpG position as hypermethylated (HM) in patients with a CEBPα locus mutation if DNA methylation level was significantly increased in the case of a CEBPα mutation (t-test, FDR ≤0.05 and Δ-value ≥0.1) and all non-hypermethylated (NHM) CpG in the case of a CEBPα mutation were classified as non-hypermethylated CpGs (t-test, FDR >0.05 and |Δ-value| <0.1). As a result, we obtained 11955 HM and 261433 NHM CpGs.
As suggested in a previous study22, unpaired RNA nucleotides are more likely to form triplexes with DNA. We predicted RNA secondary structure using RNAplfold (V 2.4.14), from the Vienna suite using a cut-off for pairing probability (-c) of 0.123,24. To search for potential interactions between ecCEBPα and DNA target regions we used only unpaired nucleotides, while the nucleotides predicted to pair were replaced with ‘N’.
DNA target regions were defined as 100 nucleotides centered at each CpG. To predict ecCEBPα triplex formation with DNA target regions, we used Triplexator (V 1.3.2)25, since it has higher accuracy of prediction14, with the following optimization parameters suggested in 22: minimum length = 10 nucleotides, error rate = 20%, G-C content = 70%, and filter-repeats = off. Using these parameters out of 307796 unique CpG loci, we predicted 272131 loci with at least one triplex and 35715 without any. Among them, 10351 and 222105 potential triplex targets were predicted in the HM and NHM regions respectively.
To estimate the statistical significance of predicted triplexes we used Triplex domain finder (TDF v 0.12.3), which clusters RNA triplex-forming oligonucleotide (TFO) into DNA binding domains (DBD)26. Briefly, all 272131 CpG loci with at least one predicted triplex were taken as input target regions. By predicting triplexes in the background regions, TDF is capable of estimating the statistical significance of ecCEBPα binding between target regions and other non-target CpGs regions. Since TDF allows only to mask regions in the genomic background rather than to select the background explicitly we had to prepare a special mask for the non-target regions. To do so we removed 35715 CpG loci with zero triplex predictions from the human genome using BEDtools subtract (BEDTools v2.29.2). TDF was implemented with a minimum triplex length (-l) of 10 nucleotides, an error rate (-e) of 20%, and (-f) to mask background loci in 100 random samplings (-n).
To validate the predicted interactions we used RNA:chromatin interactome obtained with iMARGI method capturing chromatin-associated RNA (caRNA) and their genomic interaction loci27. The data was downloaded from GEO (GSM3478205). The iMARGI dataset was mapped to the hg38 genome assembly. We used UCSC Liftover to convert ecCEBPα sequence coordinates from hg19 to hg38 sequences28. We expanded the DNA coordinates of CpGs by 3.0kb nucleotides upstream and downstream. IntersectBed from BEDTools was used to check the co-location of predicted triplexes and experimentally validated interactions of ecCEBPα29. Fisher’s exact test was calculated for the number of confirmed ecCEBPα interactions between TDF and iMARGI data.
Finally, since Illumina 450K array probes are located close to genes, we performed functional enrichment using BiNGO (v 3.0.3) (binomial test)30 to infer the biological significance of the genes potentially affected by ecCEBPα binding.
All statistical analyses were performed using R 4.0 or SciPy v1.5.1 library. Visualization was done in Cytoscape 3.2.031 and Python 3.7. Code is available at https://zenodo.org/record/438525932.
The current study explores the potential of the lncRNA ecCEBPα in the modulation of global CpG methylation status in trans via direct interaction with DNA regions. ecCEBPα (extra coding CEBPα), reported in work by Di Russio et al.20, is located on chromosome 19 and transcribed from the CEBPα locus (Figure 1a).
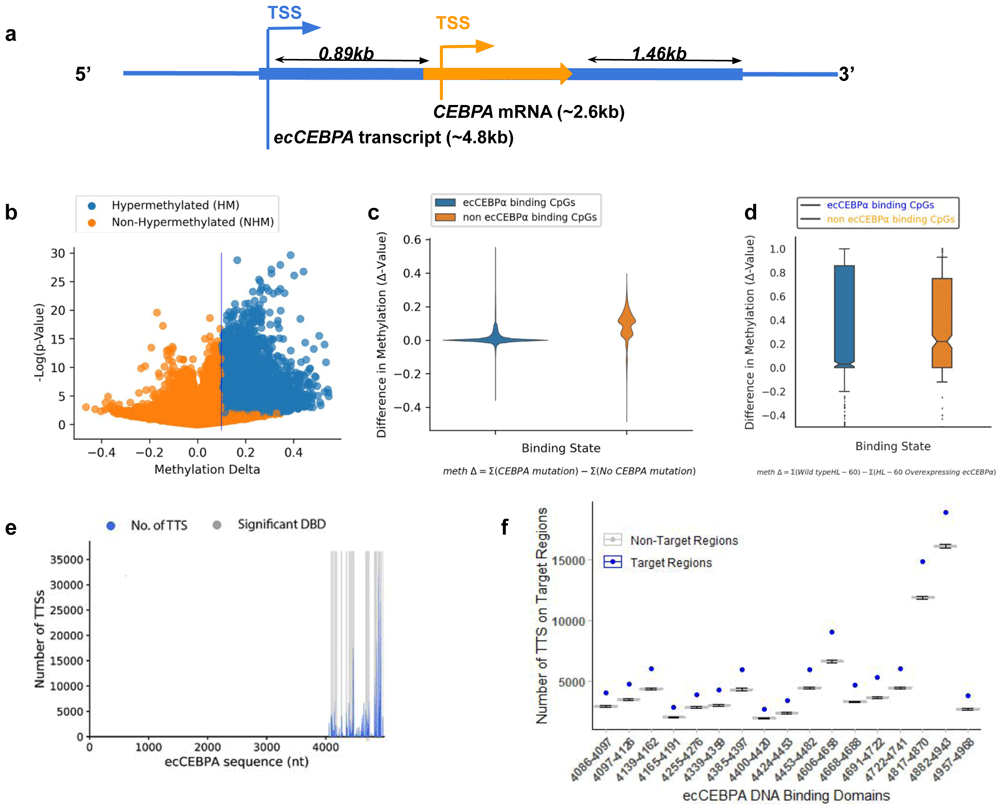
(a) Schematic diagram for transcriptional products in the CEBPα locus; CEBPα is represented by an orange arrow and ecCEBPα is represented by the blue line. (b) Global change in DNA methylation. (c) Difference in DNA methylation levels between patients with and without CEBPα mutation in the regions of ecCEBPα predicted binding (non parametric t-test, p-value >1E-10). (d) Number of DNA Triplex Target Site (y-axis) and a location of the corresponding TFO on the ecCEBPα (TFO: RNA; x-axis). (e) Number of Triplex Target sites per TFO predicted for NHM and HM CpG regions.
Mutations in CEBPα locus are a common feature of AML leading to whole genome hypermethylation (Figure 1b). Since TCGA is focused on protein-coding genes, all reported mutations are located within the CEBPα gene and could affect both CEBPα and ecCEBPα. To investigate if ecCEBPα could affect DNA methylation in trans, first we checked if it is capable of interaction via forming triple helices (triplexes) with its binding sites and if such interactions affect DNA methylation. We observed that regions capable of forming triplexes with ecCEBPα remain protected from global DNA hypermethylation observed in case of a mutation in a CEBPα locus (Figure 1c, Fisher’s exact test, p-value <0.001). Furthermore, the overall methylation profile of HL-60 cells with overexpressed ecCEBPα (Wang et al.) also have a protective effect on ecCEBPA’s binding sites in comparison to the rest of the genomic CpGs (Figure 1d, Fisher Test: p-val = 1.24E-09). This result suggests a negative relationship between DNMT access to promoter sites and ecCEBPα binding.
To investigate deeper the potential of ecCEBPα to form triplexes we used Triplex Domain Finder (TDF) - a triplex prediction tool that refines the resolution of predicted TFOs in RNA into DNA binding domains (DBD) and calculates the significance of the number of predicted triplexes for each DBD. Overall, 17 significant DBDs were identified within ecCEBPα, interspaced between sequences 4086 and 4968 towards the 3’ end of the lncRNA (Figure 1e). These DBDs form triplexes with the majority of regions protected from hypermethylation in patients with a CEBPα mutation (Figure 1f, Extended data: Supplementary Table 1). The DBD region is located downstream from the CEBPα gene suggesting that ecCEBPα binding region is not affected by the mutation in the CEBPα locus. The predicted secondary structure of the ecCEBPα sequence showed that more than 95% of sequence positions from 4087-4987 (~0.5kb from CEBPα TES) (Figure 1e) were unpaired and potentially capable of forming triple helices with the target DNA region (Figure 2a).

(a) Predicted secondary structure of ecCEBPα. Nucleotide color represents base pairing probabilities as predicted by RNAplfold. (b) Experimentally validated ecCEBPα triplexes per chromosome. The x-axis represents the chromosome and the y-axis represents the triplex potential relative to TTS. Blue points represent all TDF predictions that are present in the iMARGI dataset. (c) Functional enrichment of genes located nearby CpG with predicted triplexes. (d) Schematic representation of ecCEBPα:DNA interactions in trans and its implication on DNA methylation. The presence of ecCEBPα inhibits DNA methylation process.
Since we use relatively relaxed thresholds for triplex prediction, we decided to validate the predicted RNA:DNA triplexes using experimental data obtained with the iMARGI method, allowing detection of RNA-chromatin interactions. We identified 157 ecCEBPα contacts within the iMARGI dataset and 29 of them contained predicted triplexes. Altogether, these 29 iMARGI interactions were made up of 182 predicted TTS (Fisher’s exact test, p-value < 2.2E-16) located in cis and in trans on 14 chromosomes (Figure 2b). Chromosomes 19 (the native chromosome for ecCEBPα) are accounted for by all predictions. Since these ecCEBPA contacts have been experimentally validated, we suggested that they are the most reliable regions where ecCEBPA might have a protective effect from DNA methylation. Overall, a mean methylation delta score of experimentally validated binding sites is significantly lower than that of the non-binding sites (p-value < 1E-09) (Figure 2c).
We performed gene ontology analysis on the genes located nearby 182 ecCEBPα triplexes supported by iMARGI. Key gene ontology categories such as nucleic acid binding and transcription factor activities were enriched among putative ecCEBPα targets (Figure 2c). Representative genes among transcription factors include MLF2, SUV39H2, RBM5, UBTF, and among sequence-specific DNA binding proteins include POU2F2, MED12L, and DNASE1L1. The enrichment in transcription factors (TF) suggests that triplex formation may represent a possible mechanism employed by ecCEBPα to regulate TF methylation and as a consequence, their expression. Furthermore, out of the 33 genes we identified to be targets of ecCEBPA, 16 genes (ICAM1, PDXK, etc.) are clearly related to hematopoiesis and various leukemias. A summary of the genes and their function is provided in Table 1.
| Gene | Role | Reference |
|---|---|---|
| FLVCR1 | Alternative splicing disrupts of FLVCR1 disrupts erythropoiesis in Diamond-Blackfan anemia. (Rey et al. 2008) | https://haematologica.org/article/view/5065 |
| PSMC6 | Identified as part of a novel cluster for classifying AML risk and predicting outcomes. (Wilson et al. 2006) | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1895492/ |
| CMIP | Identified as one of the top genes expressed after rhIGFBP7 treatment (to suppress clonal growth) of AML cases. (Verhagen et al. 2018) | https://www.cell.com/cell-reports/pdfExtended/S2211-1247(18)31838-2 |
| GNA13 | A crucial signalling component of the GPR84/Beta-Catenin Signaling Axis in AML Stem Cells. (Dietrich et al. 2014) | https://ashpublications.org/blood/article/124/21/3577/97803/GNA13-a-Novel-Component-of-the-GPR84-Beta-Catenin |
| ICAM1 | Responsible for migration and adhesion of myeloid cells in hyperleukocytic AML. (Zhang et al. 2006) | https://onlinelibrary.wiley.com/doi/full/10.1111/j.1365-2257.2006.00784.x |
| PDE4C | Its mutation/absence leads to suppression of apoptotitc response in myelogenous symptoms. (Lerner and Epstein 2006) | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1383661/ |
| ANKRD27 | Identified as part of a novel cluster of competing endogenous RNA that predict AML survival and prognosis. (Wang et al. 2020) | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7432272/ |
| PDXK | Responsible for Vitamin B6 addiction of AML cells. (Chen et al. 2020) | https://pubmed.ncbi.nlm.nih.gov/31935373/ |
| NLRP3 | The NLRP3 inflammasome is upregulated as part of the stress response in AML cells. (Jia et al. 2017) | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5754918/ |
| PNPLA3 | Responsible for elevated transaminases in lymphoblastic anaemia. (Bruschi et al. 2017) | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5683790/ |
| CELSR1 | Potentially contains mutations that contribute to patient specific mutations that are responsible for the origination of AML.(Skoczen et al. 2019) | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3407563/ |
| PFKFB4 | Responsible for survival of acute monocytic myeloid leukemia by suppression of apoptosis. (Wang et al. 2020) | https://pubmed.ncbi.nlm.nih.gov/32299611/ |
| COL7A1 | Contains somatic variations that are described to be potentially pathogenic in ALL patients. (Skoczen et al. 2019) | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6978700/ |
| MED12L | Regulates hematopoietic stem cell (HSC) specific enhancers. It loss leads to loss of HSC stemness. (Aranda-Orgilles et al. 2016) | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5268820/ |
| GALNTL5 | Contains deleterious deletions that lead to copy number alteration in core binding factors of both adult and pediatric AMLs. (Kühn et al. 2012) | https://ashpublications.org/blood/article/119/10/e67/29576/High-resolution-genomic-profiling-of-adult-and |
| TAZ | Controls AML stemness and differentiation by modulating toll-like receptor (TLR) signaling. (Seneviratne et al. 2019) | https://pubmed.ncbi.nlm.nih.gov/30930145/ |
Unlike other forms of cancers, AML progression is often mutation-independent but may be explained by altered epigenetic regulation, DNA methylation specifically1,8. In this study, we elucidate a putative mechanism for the regulation of DNA methylation by ecCEBPα. In a previous study, ecCEBPα, which accompanies the transcription of CEBPα on the same locus, was shown to protect the promoter of CEBPα from DNA methylation leading to active expression20. We speculated that ecCEBPα might perform a similar function in trans. We demonstrated that ecCEBPα-DNA triplex formation might provide the molecular basis of this interaction. ecCEBPα binding presumably protects the region from genome-wide hypermethylation induced by CEBPα mutation in AML patients. Di ruscio et al. confirmed that the most mutations in CEBPα do not influence the expression of ecCEBPα but rather, its ability of the RNA to fold properly. We suggest that the inability to fold properly even in overexpressed cases may affect its structural ability to bind its targets. ecCEBPα contains a TFO/DBD-rich region at its 3’ end, with low pairing probability, suggesting that it is capable of triplex formation. Several of the predicted ecCEBPα binding sites, which include transcription factors such as MLF2, SUV39H2, RBM5, UBTF, and sequence-specific DNA binding proteins include POU2F2, MED12L, and DNASE1L1, were supported by experimental iMARGI RNA:chromatin interactions data.
Currently, the understanding of lncRNA function and mechanisms of action is limited to a few dozen well-annotated lncRNA transcripts. A few functional characterization attempts are based on the ‘guilt by association’ hypothesis, which may not resonate well with the ability of lncRNA to interact in trans33. As thoroughly reviewed previously, lncRNAs such as ecCEBPα, Dum, Dali, Dacor1, and LINCRNA-P21 interact with DNA in trans to regulate DNA methylation34. The results presented herein further demonstrate that triplex formation between ecCEBPα and CpG containing DNA regions could indeed be regulatory and protect CpG sites from DNMT activity.
Unfortunately, RNA:chromatin interaction protocols are relatively new and the data is available only for a few cell types. Since RNA:chromatin interactions are highly cell-type specific35 and lowly expressed, it is not surprising that we could validate only a few of the predicted interactions. Nevertheless, based on our results, we suggest a model of potential ecCEBPα chromatin interaction in trans (Figure 2e). In this model, ecCEBPα uses its unpaired regions to directly bind to specific DNA sequences by forming triplexes and in this way prevents DNA methylation in the region of binding. ecCEBPα binding to distant regions could be mediated either by 3-dimensional chromatin organization17 which brings them close to ecCEBPα.
Recent studies have observed that promoter or transcription start sites (TSS) regions, which tend to be rich in CpG dinucleotides, are TTS-rich and potential triplex-forming hotspots36,37. Through functional enrichment analysis, we observed that transcription factors might be preferential targets of ecCEBPα. Interestingly, previous studies have shown that the suppression of a myeloid leukemia factor (MLF2), an oncogene in breast cancer and myeloid leukemia38,39 as well as UBTF which controls rDNA expression40,41 contributes significantly to cancers upon promoter hypermethylation40,42. The suppressor of variegation 3-9 homolog 2 (SUV39H2), a histone-lysine-N-methyltransferase which regulates the hypermethylation H3K9 has also been reported to indirectly influence over 450 promoters in AML43. Having in mind that ecCEBPα is transcribed from CEBPα locus - a key transcription factor of hematopoiesis - this lncRNA could participate in the formation of a hub in the hematopoiesis regulatory network.
In conclusion, we have shown that ecCEBPα could serve as a trans-acting regulatory agent protecting its binding sites from genome-wide CpG methylation, and its dysregulation could contribute to aberrant methylation profile in AML patients. These results suggest a novel regulatory mechanism for ecCEBPα as a modulator of DNA methylation through triplex formation providing a foundation for sequence-specific engineering of RNA for regulating methylation of specific genes.
Complete ecCEBPα sequence retrieved from the human genome (hg19, chr19: 33298573-33303358): https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.13/
CEBPα mutation data for 186 AML patients retrieved from the Cancer Genome Atlas (TCGA): http://firebrowse.org.
GEO: Embryonic kidney that expresses SV40 large T antigen, Accession number GSM3478205: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM3478205
GEO: DNA Methylation by Reduced Representation Bisulfite Seq from ENCODE/HudsonAlpha GSM980576: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM980576
Zenodo: josoga2/eccebp-alpha-project: F1000 Code Release, https://doi.org/10.5281/zenodo.438525932
This project contains the following underlying data:
Code used for analysis available from: https://github.com/josoga2/eccebp-alpha-project/tree/f1000
Archived code as at time of publication: https://doi.org/10.5281/zenodo.438525932
Zenodo: Supplementary Data for Secondary structure and DNA binding domain prediction, http://doi.org/10.5281/zenodo.443322244.
This project contains the following extended data:
Supplementary Table 1: Summary table of DNA binding domains (DBD), the counts of target regions within the genome and statistical analysis.
ecCEBPα secondary structure prediction with RNAplfold
Data and code are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (1)