Keywords
SARS-CoV-2, COVID-19, Mutation time maps, GISAID, Interactive SVG
This article is included in the Pathogens gateway.
This article is included in the Emerging Diseases and Outbreaks gateway.
This article is included in the Bioinformatics gateway.
This article is included in the Coronavirus (COVID-19) collection.
SARS-CoV-2, COVID-19, Mutation time maps, GISAID, Interactive SVG
In the last few weeks of 2020, new severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) mutations in the United Kingdom (UK) were reported1. Although coronavirus genome mutations have been previously discovered and announced throughout the year, including the widely discussed D614G missense change in the spike protein2,3, the latest recurring surface protein mutations to be identified (e.g. N501Y, P681H) are cause for concern. The SARS-CoV-2 viral S gene encodes a surface glycoprotein, which upon interaction with host ACE-2 receptors, makes it possible for the coronavirus to gain entry to host cells and propagate. The reported changes to its sequence may be associated with increased virulence4, infectivity3 and overall fitness5. The global response to those recent reports has been swift, with several countries shutting down air travel from the UK. This highlights the severity of the situation and the importance to track genomic variations and their predicted effects over time and space.
The rapid evolution of the SARS-CoV-2 genome in human hosts has prompted us to map all nucleotide changes that have appeared in 2020, since the first genome sequence of a COVID-19 patient isolate from the outbreak epicentre in Wuhan, China was made public6. For this, we leveraged the collaborative efforts of hundreds of institutions worldwide who have graciously shared over 260,000 SARS-CoV-2 genome sequences with the GISAID central repository since early January 20207. Our mutation time maps show the staggering number of nucleotide variants that have accumulated on the whole viral genome throughout the year, and especially since fall 2020, and in the six most populated continents. Here we present key features of these maps and how they may be of utility to researchers.
We first downloaded all complete, high-coverage SARS- CoV-2 genomes from GISAID7 on January 23rd, 2021 (human hosts samples collected). We then ran a genome polishing pipeline, which consists of ntHits8 (v0.1.0 -b 36 -outbloom -c 1 -p seq -k 25) followed by ntEdit9 (v1.3.4 -i 5 -d 5 -m 1 -r seq_k25.bf) and required at most 0.5 GB RAM and executed in ~1 sec. per genome on a single CPU. We used the first published SARS-CoV-2 genome isolate6 (WH- Human 1 coronavirus, GenBank accession: MN908947.3) as the reference and each individual GISAID genome in turn as source of kmers to identify base variation relative to the former. The variant call format (VCF) output files from ntEdit were parsed and we tallied, for each submitted GISAID genome, the complete list of nucleotide variations. We next organized each nucleotide variant by sample collection date, continent of origin and, when applicable, evaluated its effect on the gene product that harbours the change to output an interactive scalable vector graphics (SVG) file. The script we developed to generate the maps is written in PERL and distributed under GPLv3. Users wishing to generate custom maps can download the script from Zenodo10.
We analyzed nucleotide variations over time in over 260,000 SARS-CoV-2 viral genomes, submitted to the GISAID initiative7 from around the globe, relative to that of the ground zero COVID-19 clinical isolate6. We mapped each mutation that was observed in five or more genomes each day. The 2020 calendar year from January 1st 2020 (day 1) to December 31st 2020 (day 366) is organized in a circle where each radius represents a day (1 day = 0.98 degree) and data points represent mutations along the reference genome sequence from 1 (closest to center) to 29,903 bp (near the outer rim). The size of each point is in log10 scale of the number of contributing viral genomes collected on that day that has the mutation, with colour assignments indicating the continent of origin where the mutation is observed. A mouse over each data point reveals the collection date, the nucleotide variant, the continent and associated number of contributing genome sequences (including daily sample fraction) and, when applicable, the gene product and predicted amino acid change.
From the SARS-CoV-2 genome mutation time map (Figure 1A), we observe the first persistent mutations (≥5 genomes/day) appearing in late February 2020, including the prevalent D614G mutation in Europe on February 22nd (albeit since January in fewer samples, Figure 1B). From there, the original coronavirus genome sustained many changes overtime (5,468 distinct variants mapped in 2020 as of January 23rd, 2021), including a sizeable proportion (56.8 %) of missense mutations. It is immediately evident from Figure 1A that variations from Europe account for a larger share (71.2%) of the variants mapped. Further, there appears to be a surge in variations identified in late summer/throughout fall 2020 in this continent. This may be explained by a disproportionate number of submissions with samples originating from this jurisdiction as the second wave hit hard. Thus, caution in interpreting the map is warranted. Of note, the spike protein gene variant N501Y, observed on our maps in the UK in late September 2020 (Figure 1), is consistent with an earlier study reporting on its recurrent emergence within this time frame1. We think these maps will be of utility to researchers in their exploration of SARS-CoV-2 mutations and their predicted effect over time.
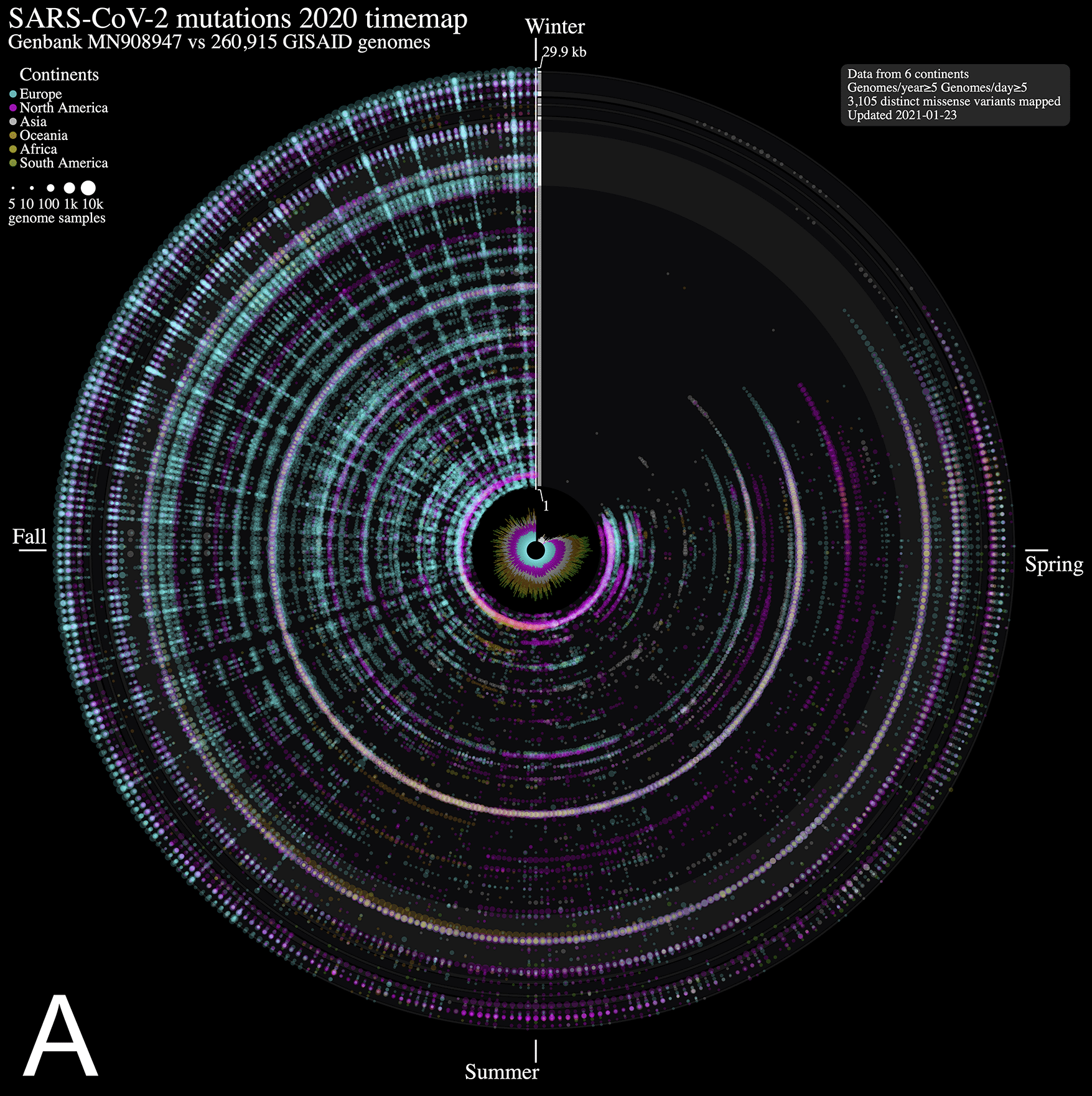
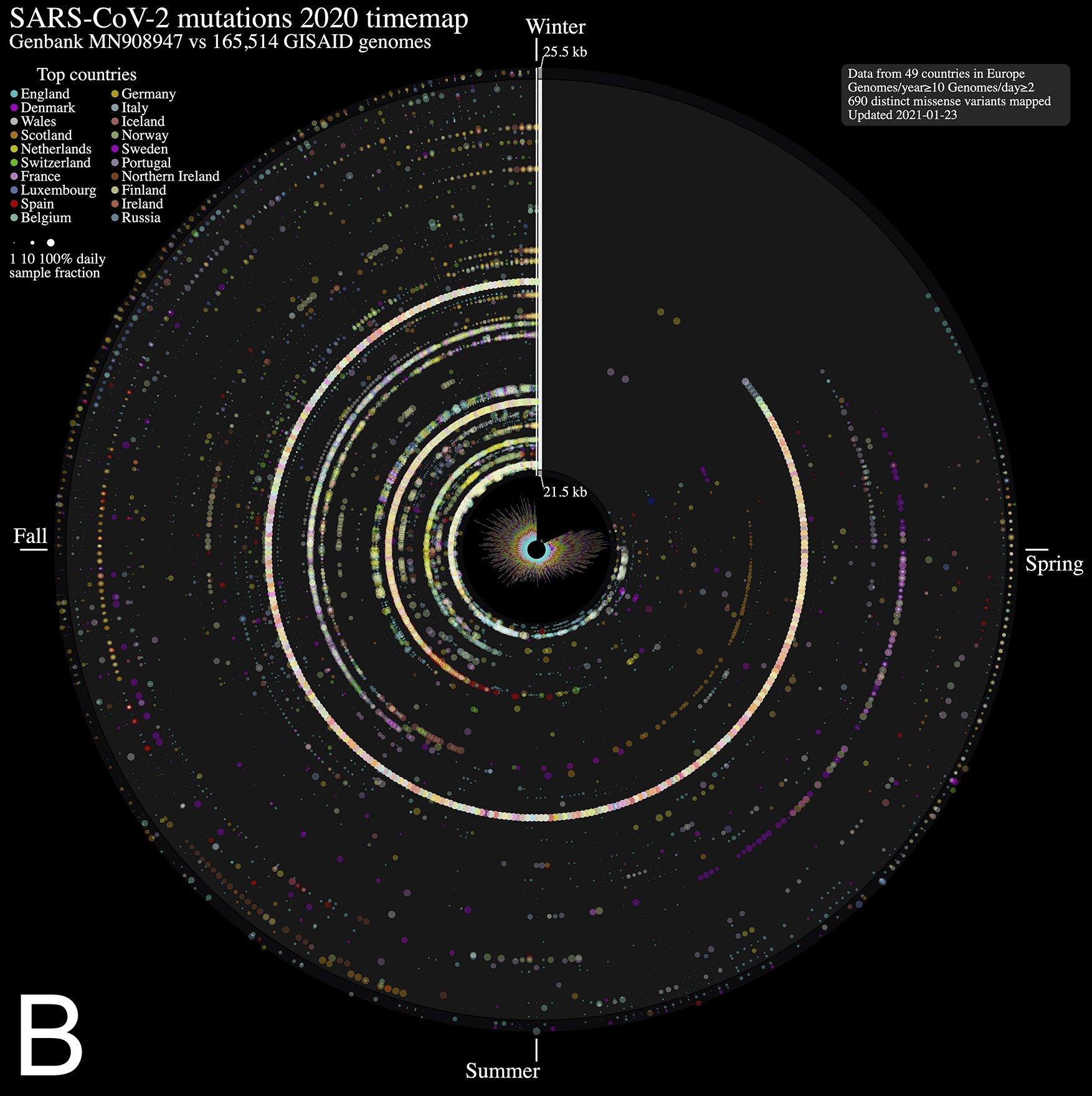
ntEdit was used to map nucleotide variations between the first published coronavirus isolate from Wuhan, China in early January and over 260,000 SARS-CoV-2 genomes sampled from around the globe during the 2020 coronavirus disease 2019 (COVID-19) pandemic. The maps show missense mutations arising daily (A) in the world within the whole viral genome, with the reference genome represented by the vertical axis from bases 1 to 29.9 kbp and (B) in Europe within the spike protein gene. Alternating dark/light grey vertical rectangles and associated tracks depict, starting from the center, SARS-CoV-2 genes orf1ab, S, ORF3a, E, M, ORF6, ORF7a, ORF8, N, and ORF10. Mutations identified daily are represented by circles in a given radius and are coloured by regions and sized relative to raw count (panel A) or ratio (panel B) of the daily samples. A stacked bar plot (center) shows sample count. The 2020 calendar year mutations are organized clockwise from the upper vertical. Hovering the mouse cursor over each data point reveals additional insights (not shown).
The SARS-CoV-2 genome sequences can be accessed via the GISAID central repository. Processed single nucleotide variant (SNV) data is available from https://www.bcgsc.ca/downloads/btl/SARS-CoV-2/mutations/.
- Maps are available from: https://bcgsc.github.io/SARS2
- SNV detection source code is available from: https://github.com/bcgsc/ntedit
- Archived source code at time of publication: https://doi.org/10.5281/zenodo.446984010
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)