Keywords
Visualization, Gene expression, Ontology, o²S²PARC
This article is included in the Genomics and Genetics gateway.
This article is included in the Bioinformatics gateway.
Visualization, Gene expression, Ontology, o²S²PARC
Transcriptome data has been used to understand the local microenvironment, molecular signals, and cell-cell interaction in cells, tissues, and organs in multiple diseases, such as Alzheimer’s disease,1 Parkinson’s disease,2 and much more. In this study, we focus on the gene expression data, particularly the differentially expressed genes (DEGs) and their associated ontologies: (i) the cellular component (CC) that describes the subcellular structures and macromolecular complexes, often used to annotate cellular locations of gene products; (ii) the biological process (BP) that describes the biological programs consisting of multiple molecular activities, such as DNA repair or signal transduction; (iii) and the molecular function (MF) that describes molecular-level activities performed by gene products, such as “catalysis” or “transport”.
This study was performed during the SPARC FAIR Codeathon in August 2022 organized by the National Institute of Health (NIH) SPARC program. We developed a gene expression data visualization tool created and published on the o2S2PARC, Open Online Simulations for Stimulating Peripheral Activity to Relieve Conditions platform, a simulation and analysis platform aiming to initially perform interactive peripheral nerve system neuromodulation/stimulations and to visualize its physiological impact on organs.3 However, the platform currently hosts tools for multiple biological and physiological analyses but does not provide a tool for transcriptomics and gene expression data analysis or visualization.
The tool was initially created to visualize the transcriptomics data on the SPARC Portal platform, a fully open-accessible database consisting of a wide variety of datasets across multiple scales, such as organs, species, and datatypes.4 The platform is created and maintained by the Stimulating Peripheral Activity to Relieve Conditions (SPARC) program, funded by NIH. It was initiated to advance the understanding of nerve-organ interactions and to expedite the invention of therapeutic medicine and devices that modulate electrical activity in nerves to promote organ function.5 The SPARC consortium runs under the FAIR data sharing policy (consists of the principle of Findable, Accessible, Interoperable, and Reusable), according to the SPARC Data Structure (SDS).
Implementation
The tool is created as a template in the o2S2PARC platform. The platform is accessible on all web browsers. It requires pandas 1.4.3, bioinfokit 2.0.8, numpy 1.22.1, matplotlib 3.5.2, seaborn 0.11.2, and goatools 1.2.3. All the requirements are integrated within the tool and automatically installed.
Operation
The tool includes two pipelines encoded in two separate python jupyterlab notebooks. The first pipeline identifies the DEGs based on the p-value, set to p-value < 0.05 as default, and determines the expression profile of the genes:
• p-value > 0.05: “Not differentially expressed”
• p-value < 0.05 and the LogFC value > 0: “Upregulated”
• p-value < 0.05 and the LogFC value < 0: “Downregulated”
The pipeline also performs the ontology analysis for the differentially expressed genes, to determine the cellular components, biological processes, and molecular functions associated with these genes. The Biological processes, molecular functions, and cellular component ontologies are represented in six similar separate Barplots, as represented in Figure 1.
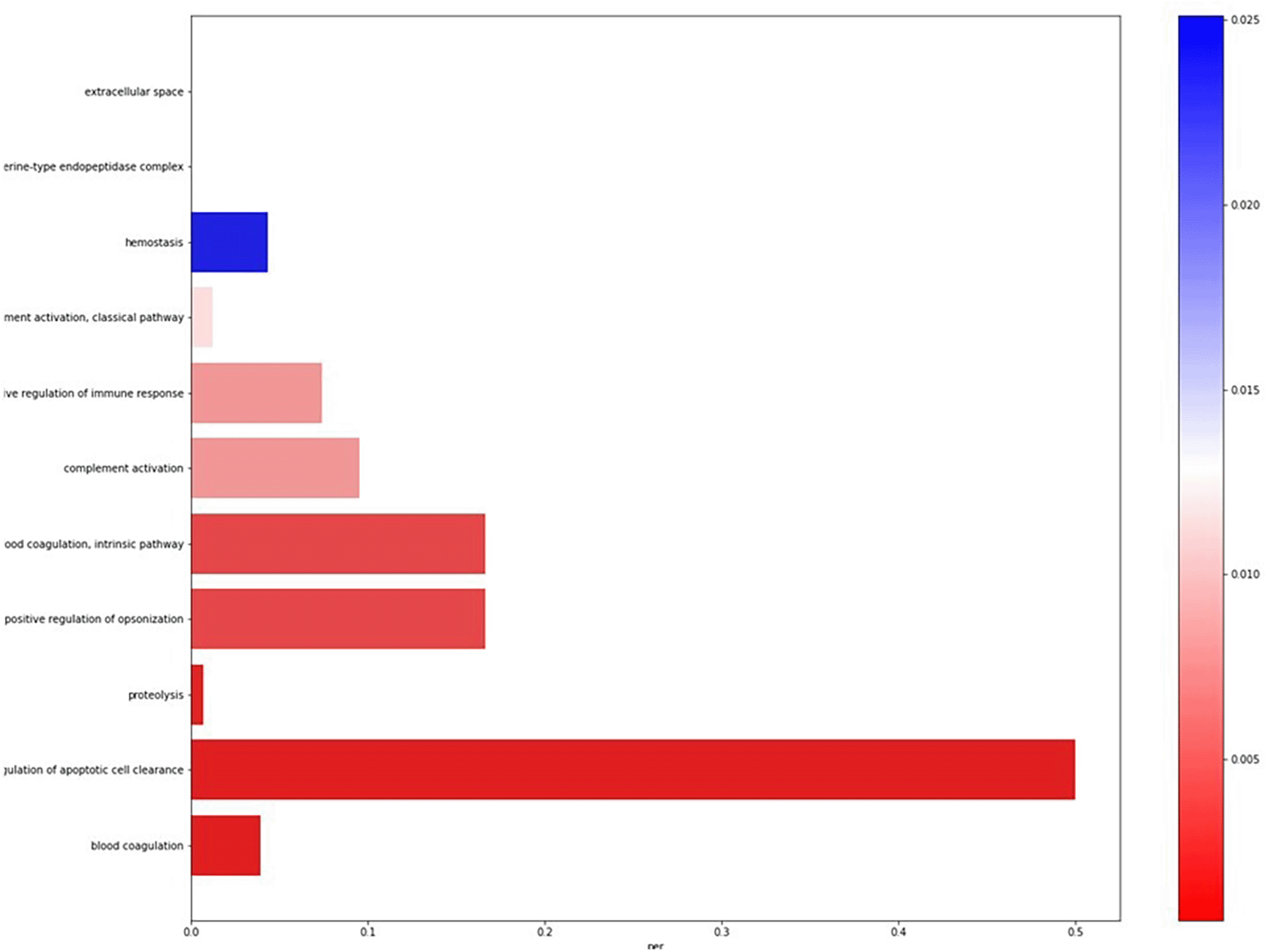
The genes-related ontology data were downloaded from the NCBI database (https://www.ncbi.nlm.nih.gov/). The file for the human species is provided as default. The user needs to provide a file as input if the transcriptomics data correspond to other species.
The second pipeline takes two CSV files as input. The gene’s expression profile is determined, as in the first pipeline, for the two datasets. The common and uncommon genes count is performed. And the gene expression profiles in the two datasets are compiled in a single csv file for further analysis.
A web browser extension was developed, using HTML and CSS programming, as a user guide. The extension is helpful for the new SPARC platform users. It guides the user step by step from downloading transcriptomics data from the SPARC portal database, providing a raw data analysis workflow, and explaining the “Gene expression data visualization” tool.
The tool was initially created to visualize the SPARC Portal platform transcriptomics data. However, it could be used to visualize any expression data csv file. The pipeline validation was performed using two datasets from the Gene Expression Omnibus (GEO) database6 corresponding to the early and advanced stages of multiple sclerosis disease (MS) in human patients (GSE 126802 and GSE 10800).
The early-stage dataset GSE1268027 provides microarray gene expression analysis raw data from the subcortical normal-appearing white matter from 18 MS donors and the white matter of 9 control donors. The advanced stage dataset GSE1080008 provides microarray gene expression data from 7 chronic active MS demyelinated lesions, 8 inactive MS lesions, and white matter of 10 control donors.
The tool was used to visualize the first dataset data, to determine the genes and pathways implicated in the occurrence of the disease. Then we compared the two datasets to determine the genes and pathways implicated in the disease progression.
The tool includes two pipelines, one to visualize the expression data from a single csv file, and the second to compare two datasets.
The dataset expression data are visualized in a volcano plot format, as represented in Figure 2.
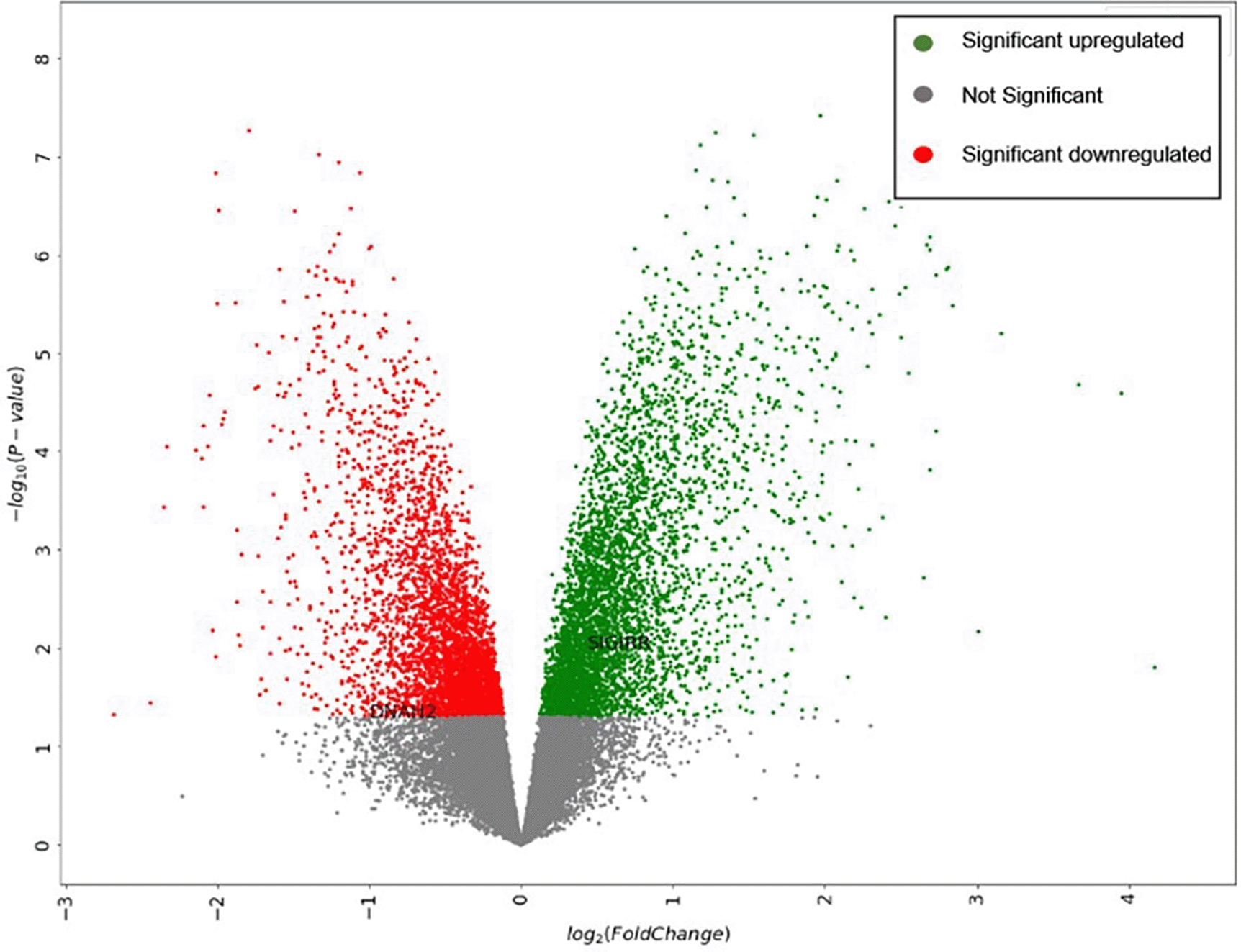
The pipeline also determines the ontologies associated with the DEGs: (i) BP associated with upregulated genes; (ii) MF associated with upregulated genes; (iii) CC associated with upregulated genes; (iv) BP associated with downregulated genes; (v) MF associated with downregulated genes; and (vi) CC associated with downregulated genes. The top ontologies are represented in six barplots.
The second pipeline determines the genes with similar expression profiles in the two datasets and most importantly those with different profiles, which is useful to compare two cells, tissues, or diseases.
It also generates a table resuming the gene’s count, as represented in Table 1.
| data2_expression | Downregulated | Not differentially expressed | Upregulated |
|---|---|---|---|
| data1_expression | |||
| Downregulated | 834 | 1048 | 82 |
| Not differentially expressed | 6850 | 35307 | 5292 |
| Upregulated | 47 | 792 | 415 |
The tool is publicly available for all the o2S2PARC platform users. And the user guide Browser extension is available in the project repository (https://github.com/SPARC-FAIR-Codeathon/Transcriptomic_oSPARC).
There are multiple transcriptomic datasets available on the SPARC Portal,9 containing a wide range of species from humans, pigs, mice to rats; anatomical structures include neurons for multiple organs and physiological systems; analysis methods involve RNA sequencing, real-time PCR; small molecule FISH (RNAscope) probes, and multiple others. The SPARC portal datasets provide a wide variety of transcriptomic data, however, there is no automatic gene expression data processing or visualization tool on the SPARC system.
Transcriptomics has been increasingly favored by researchers and clinicians in prioritizing specific systems and networks,2 finding biomarkers,10 developing precision medicine strategies,10 monitoring disease progressions, and predicting treatment effects.11
The tool is useful in helping transform the transcriptome data into visualizable DEGs and gene ontology (GO) analysis in a one-step standardized format. Nowadays, DEGs and GO are commonly utilized tools in detecting potential key pathways, molecules, and cells related to target tissues, organs, and diseases.10–13
The “Gene expression data visualization” tool represents a fast and easy online visualization tool, that does not require any coding skills, to identify the gene expression changes among any target objects, such as species, tissues, and diseases. The browser extension represents an easy and detailed guide for the whole procedure. Currently, the tool requires processed data files as input. Future versions could include the expression raw data analysis.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)