Keywords
apoptosis, cancer, cell cycle, cyclins, H522 cells, lung adenocarcinoma, spiroleucettadine
This article is included in the Cell & Molecular Biology gateway.
This article is included in the Oncology gateway.
apoptosis, cancer, cell cycle, cyclins, H522 cells, lung adenocarcinoma, spiroleucettadine
Lung cancer is divided broadly into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). NSCLC accounts for 80–85% of lung cancer deaths (Navada, Lai et al., 2006, Lemjabbar-Alaoui, Hassan et al., 2015) and is one of the leading causes of death worldwide (Pao and Girard, 2011, Ettinger, Akerley et al., 2013). NSCLC can be further divided into squamous cell lung cancer (~25-30% of lung cancers), adenocarcinomas (~40% of lung cancers) or large cell anaplastic carcinomas (~10% of lung cancers) (Lemjabbar-Alaoui, Hassan et al., 2015). Adenocarcinomas arise from alveolar cells situated in the smaller airway epithelium (Travis, Brambilla et al., 2015). Approximately 30-60% of lung adenocarcinomas arise through the development of mutations in epidermal growth factor receptor (EGFR), Kirsten rat sarcoma viral oncogene homolog (KRAS) and anaplastic lymphoma kinase receptor (ALK) genes (Herbst, Heymach et al., 2008, Bronte, Rizzo et al., 2010, Pao and Girard, 2011) with a small percentage containing other targetable oncogenic drivers (Liu, Du et al., 2020). Of the large percentage of lung adenocarcinoma patients with no targetable mutations, only approximately 20% will show even a temporary and partial response to existing chemotherapies, although a prolonged survival can still occasionally occur (Nishio, Nakamura et al., 1999).
Although there has been considerable progress in the treatment of NSCLC in targeted drug treatment and immunotherapy, there has been relatively little progress in the development of new chemotherapies, particularly in comparison to other cancers (Lemjabbar-Alaoui, Hassan et al., 2015). As a minority of NSCLCs have a molecular profile suitable for treatment with targeted or immunotherapies, this means that there remains an unmet need for more chemotherapies for NSCLC.
(-)- Spiroleucettadine is an alkaloid first isolated by Ralifo and Crews (2004) from the yellow Leucetta sea sponge. Following the synthesis of spiroleucettadine by Lamb, Aberle et al. (2017), spiroleucettadine was tested for anti-cancer effects against the NCI-60 cell lines (Badart, Barnes et al., 2020). Spiroleucettadine inhibited the growth of multiple cancer types including leukaemia, melanoma, non-small cell lung, colon, renal and ovarian cancers with varying growth inhibition. Of particular interest, spiroleucettadine had the highest anti-proliferative activity against the NSCLC cell line H522 with an EC50 of 0.37 μM (Badart, Barnes et al., 2020).
The mechanism of action for these anti-proliferative effects has not been determined. In this study we investigated the changes in cell death and cell proliferation in response to spiroleucettadine, as well as changes in mediators of apoptosis and the cell cycle to characterise the late mediators of cancer cell growth suppression by spiroleucettadine, toward the aim of ultimately determining specific cellular target(s) leading to these effects.
We performed our initial experiments on three human lung adenocarcinoma cell lines, each with distinct oncogenic drivers: H522 (p53 mutated – gifted from Eccles Lab, University of Otago, NZ); A549 (KRAS mutated – gifted from Giles Lab, University of Otago, NZ); and H3122 (ALK mutated – gifted from Engelman Lab, Harvard Massachusetts General Hospital Cancer Center, US). As H522 cells showed the greatest cytotoxic response to spiroleucettadine we performed our later experiments investigating mechanisms of action only on these cells.
(-)-Spiroleucettadine was prepared using the procedures described by Badart et al. (2020). Purity was ascertained using high performance liquid chromatography (HPLC) and found to be >98% pure and > 96% enatiomenic excess (ee). HPLC conditions included: Diacel CHIRALPAK IC-3 chiral column, Shimadzu ELSD-LT II detector, eluting with acetonitrile.
Colourless semi-solid
[α]20D = -23.0 (c = 1.23, CHCl3)
1H NMR (500 MHz, MeOH-d4) δ: 7.45 (d, J = 8.6 Hz, 2H), 6.99 (dd, J = 10.5, 2.2 Hz, 1H), 6.88 (d, J = 8.5 Hz, 2H), 6.01 (app. d, J = 10.8 Hz, 1H), 5.93 (dd, J = 10.1, 2.3 Hz, 1H), 5.92 (dd, J = 10.1, 2.3 Hz, 1H), 3.80 (s, 3H), 3.21 and 3.17 (abq, J = 14.5 Hz, 2H), 2.79 (s, 3H), 2.40 (s, 3H), 2.22 and 2.07 (abq, J = 13.2 Hz, 2H).
13C NMR (125 MHz, MeOH-d4) δ: 186.9, 161.2, 160.2, 152.5, 151.5, 134.2, 129.0, 128.4, 127.4, 114.2, 104.0, 84.0, 78.6, 55.7, 49.0, 38.6, 29.0, 26.2.
νmax (ATR-IR) cm-1 3293, 3057, 2928, 2853, 2837, 2809, 1699, 1669, 1629, 1610, 1511, 1437, 1393, 1329, 1300, 1246, 1177, 1158, 1031, 1013, 733, 500, 585.
HRMS-ESI: calcd. for C20H23N3NaO4 [M + Na+]: 392.1581; found 392.1592.
H522 and H3122 cells were maintained in Roswell Park Memorial Institute (RPMI)1640 medium supplemented with 1% streptomycin/penicillin and 5% foetal bovine serum (FBS) (Life Technologies, NZ Ltd), while A549 cells were maintained in RPMI1640 supplemented with 1% streptomycin/penicillin and 2% FBS. All cell lines were incubated at 37°C, 5% CO2, 95% humidified air. Cells were passaged at approximately 80-90% confluency. Phosphate buffered saline (PBS) (Sigma-Aldrich, US) was used to wash cells before trypsinisation using TrypLE (ThermoFisher, NZ) to detach cells from the flasks. Cells were then centrifuged for three minutes at 1200 rpm in fresh media. Supernatant was removed, and cells were resuspended in fresh media for transfer into new tissue culture flasks.
H522 cells were harvested at approximately 80-90% confluency; cells were PBS-washed before trypsinisation using TrypLE to detach cells from flasks. Cells were then centrifuged for three minutes at 1200 rpm in fresh media, the supernatant removed, and cells resuspended in fresh media for plating. H522 cells were seeded into 96-well plates at a density of 10×103 cells/well and incubated for 24 hours to allow cells to adhere to the plate. Cells were then treated with increasing concentrations of spiroleucettadine (0, 0.01, 0.05, 0.1, 0.2, 0.3, 0.5, 0.75, 1, 5, 10, 50 μM) and further incubated for 72 hours, 96 or 144 hours. Following this, cells were fixed with 10% trichloroacetic acid (TCA, Sigma-Aldrich, NZ) for 30 minutes at 4°C. TCA was rinsed off using distilled water and the plate was dried. Sulforhodamine B (SRB, Sigma-Aldrich, NZ) (0.4%) in acetic acid (1%, Merck, US) was added to wells to stain for cellular protein for 10 minutes at room temperature. SRB was then aspirated, and wells were washed with 1% acetic acid and the plate was dried. Tris/HCl (10 mM, Sigma-Aldrich, NZ) was then added to solubilise the SRB dye. Absorbance of wells was read using the Benchmark Plus microplate spectrophotometer at 510 nm. Absorbance values were then used to calculate approximate cell number from a constructed standard curve of absorbance versus cell number.
H522, H3122 and A549 cells were harvested at approximately 80-90% confluency. PBS was used to wash cells before trypsinisation using TrypLE to detach cells from plastic vessels. Cells were then centrifuged for three minutes at 1200 rpm in fresh media. The supernatant was removed, and cells were resuspended in fresh media for plating. H522, H3122 and A549 cells were counted using a haemocytometer, seeded into 96-well plates at a density of 10×103, 7×103 and 4×103 cells/well, respectively, and incubated for 24 hours in order for the cells to adhere to the wells. Cells were then treated with the vehicle control (dimethyl sulfoxide, DMSO, Sigma-Aldrich, US) or increasing concentrations of spiroleucettadine (0, 0.01, 0.05, 0.1, 0.2, 0.3, 0.5, 0.75, 1, 5, 10, 50 μM for H522 cells or 0, 0.01, 0.05, 0.1, 0.2, 0.5, 1, 2, 3, 5, 10, 50, 100 μM for A549 and H3122 cells) and further incubated for 72 hours. The same procedure was conducted for the SRB stain as stated in the “Time course cell number assay” section. The concentration of each drug required to reduce cell viability by 50% (EC50) was determined from three-parameter Hill equations fit to data using nonlinear regression using GraphPad Prism 8 software, from three independent experiments, performed in triplicate. EC50 values are expressed as mean ± SD.
Cell cycle analysis was carried out using flow cytometry. H522 cells were harvested at approximately 80-90% confluency and cells were washed with PBS before trypsinisation using TrypLE to detach cells from flasks before being centrifuged for three minutes at 1200 rpm in fresh media. After the supernatant was removed, cells were resuspended in fresh media for plating at a density of 3×105 cells/well in six-well plates. Plates were then incubated for 24 hours to allow for cell adherence. Cells were then treated with either vehicle control (0.1% DMSO) or spiroleucettadine concentrations standardised to the cell viability EC50 (0.5×, 1.0× and 2.0× EC50) for 24 hours.
Medium was then removed, and cells were washed with PBS before trypsinisation using TrypLE for approximately five minutes. Fresh media was then added, and the cells were collected in falcon tubes. Cells were then centrifuged at 2000 rpm for four minutes. Supernatant was then discarded, and the pellet was resuspended in cold PBS. Centrifugation was repeated once before fixing the cells using cold 70% ethanol dropwise while gently mixing with a vortex. Tubes were stored at 4°C until analysis was carried out. On the day of flow cytometry analysis, cells were centrifuged at 1500 rpm for 10 minutes at 4°C, supernatant was discarded, and pellet resuspended in cold PBS. Centrifugation was carried out as described above and cells were resuspended in FxCycle PI/RNase solution (Life Technologies, US). The cell suspension was then placed in test tubes and incubated in the dark at room temperature for 30 minutes. Cell cycle assay was then carried out using a BD FACSCanto II flow cytometer (BDBiosciences, US). Data from three independent experiments were analysed using FlowJo analysis software.
The mode of cell death following exposure to spiroleucettadine was determined for H522 cells using propidium iodide (PI, Waltham, Massachusetts, US) and annexin V-APC (BD Biosciences, San Jose, California, US) staining by flow cytometry. The cells were harvested at approximately 80-90% confluency and PBS was used to wash cells before trypsinisation using TrypLE. Cells were then centrifuged for three minutes at 1200 rpm in fresh media. The supernatant was removed, and cells were resuspended in fresh media for plating. Plates were incubated for 24 hours to allow for cell adherence. Cells were then treated with either vehicle control (0.1% DMSO) or various concentrations of spiroleucettadine (0.5×, 1.0× and 2.0×EC50) for 24 hours.
Media was collected and cells were washed with warm PBS before trypsinisation using TrypLE for approximately five minutes. Fresh media was then added and collected in falcon tubes. Cells were then centrifuged at 2000 rpm for four minutes at 4°C. The supernatant was then discarded, and the pellet was resuspended in cold PBS. Centrifugation, resuspension, and centrifugation (as previously described) was carried out once more. The supernatant was discarded, and the cell pellet was resuspended in annexin V binding buffer (0.1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 1.4 M NaCl, 25 mM CaCl2, pH 7.4, Sigma-Aldrich, US) and transferred to a Falcon 5 mL FACS tube. Annexin V-APC was added to each tube and mixed gently prior to the 15-minute incubation in the dark at room temperature. Following this incubation, annexin V binding buffer and PI (50 μg/mL) was added to each tube and kept in the dark until analysis.
Samples were analysed on BD FACSCanto II flow cytometer. Data obtained was analysed using FlowJo analysis software (BD Biosciences). Three biological independent experiments were performed.
Primary antibodies against CDK2 (RRID:AB_22761290), CDK4 (RRID:AB_20783990), cyclin B1 (RRID: AB_491024), cyclin D1 (RRID: AB_2259616), Bax (RRID: AB_2716251), Bcl-2 (RRID:AB_2064177), Bim (RRID: AB_10692515), and cleaved caspase 3 (RRID: AB_2341188) were purchased from Cell Signalling Technology (Danvers, Massachusetts, USA). Antibodies against β-tubulin (RRID: AB_477577) and CDK1 (RRID: AB_258879) were purchased from Sigma-Aldrich (USA). Antibodies against pCDK1 (RRID: AB_2533722) and pCDK4 (RRID: AB_2809179) were purchased from Invitrogen (Waltham, Massachusetts, USA). Horseradish peroxidase (HRP) conjugated goat anti-mouse and HRP conjugated goat anti-rabbit secondary antibodies were purchased from Merck Millipore (Burlington, Massachusetts, USA).
Cell lysate preparation
H522 cells were seeded in petri dishes at a density of 1×106 cells/dish and incubated in 5% CO2, 95% humidified air, 37°C for 24 hours. H522 cells were treated with vehicle (0.05% DMSO) or spiroleucettadine (0.5 ×, 1.0 × and 2.0 × EC50) for 24 hours. On the following day, cells were washed with ice-cold PBS and lysed with lysis buffer (50 mM Tris base (pH-7.5), 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid EDTA (Merck (Kenilworth, New Jersey, USA), 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, 10 mM sodium fluoride (Sigma-Aldrich, US) and complete protease inhibitor cocktail (Sigma-Aldrich, US)). Cell lysates were collected by scraping and sonicated three times for 7 s each followed by centrifugation at 10,000 rpm for 8 min at 4°C. The supernatant was transferred to a new Eppendorf tube before protein concentration was determined. The protein concentration of cell lysates was determined by bicinchoninic acid (BCA) assay as described in Smith et al., (1985). Briefly, bovine serum albumin (BSA, Sigma-Aldrich, NZ) protein standard (0.5 mg/mL) was used to obtain the standard curve. BCA solution (50:1) was prepared freshly by adding 50 volumes of BCA protein assay reagent A (Merck (Kenilworth, US) and 1 volume of 4% copper sulphate pentahydrate (CuSO4.5H2O) (Sigma-Aldrich, US) solution. BCA solution was added to each well (containing either the standards or samples) and incubated in the dark at 37°C for 30 minutes. Absorbance was measured at 562 nm using Bio-Rad Benchmark Plus microplate reader. A standard curve was used to determine the protein concentration of samples and each sample was normalised to obtain 200 μg/μL of protein. Laemmli sample buffer (62.5 mM Tris HCl, 1% (w/v) SDS, 10% glycerol, 0.005% bromophenol blue, 355 mM β-mercaptoethanol, Sigma-Aldrich, US) (pH 6.8)) was added to each sample and denatured by boiling at 95°C for 5 min. The samples were frozen at -20°C until required.
Gel electrophoresis and antibody labelling
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate proteins based on their molecular weight (Blake, Johnston et al., 1984). Different percentages of resolving gels ranging from 10% to 15% (30% acrylamide/bisacrylamide (29:1) (Bio-Rad, US) solution, lower Tris buffer (1.5 M tris/HCl pH8.8, 0.4% SDS), 50% glycerol, 10% APS, dH2O, N,N,N,N-tetramethyethylene diamine TEMED, Sigma-Aldrich, US) and a stacking gel (30% acrylamide/bisacrylamide (29:1) solution, upper Tris buffer (0.5 M tris/HCl pH6.8, 0.4% SDS), 10% of APS, dH2O, TEMED) were made depending upon the type of proteins of interest. Samples were defrosted and 20 μL of protein was loaded in each well of gels. Precision Plus Protein kaleidoscope (a molecular weight protein marker) was loaded in each gel to identify the respective proteins based on their molecular weight. Gels were run in a Bio-Rad Mini-Protean III apparatus at 80 V in SDS-running buffer (25 mM Tris base, pH 8.3, 0.192 M glycine, 0.1% SDS) for approximately for 15 min until the protein get migrated to the end of stacking gel. Then, the voltage was increased to 120 V until the protein reached to the bottom of resolving gel.
The proteins were transferred to a methanol activated polyvinylidene fluoride (PVDF, Sigma-Aldrich, US) membrane in transfer buffer (25 mM Tris-base, pH 8.3, 0.192 M glycine, 0.1% sodium dodecyl sulphate (SDS, Sigma-Aldrich, US) and 20% methanol) using a Bio-Rad wet transfer system at 100 V for 90 minutes. After the completion of the transfer, membranes were placed in 2% BSA blocking solution and placed on a shaker for one hour at room temperature. This was followed by incubation with respective primary antibody (diluted in 2% or 5% BSA) overnight at 4°C. The dilution of primary antibody used was as follows: CDK1 (1:1000), CDK2 (1:1000), CDK4 (1:1000), CDK6 (1:1000), pCDK1 (1:1000), pCDK4 (1:1000), cyclin B1 (1:1000), cyclin D1 (1:1000), cleaved caspase-3 (1:1000), Bcl-2 (1:1000), Bim (1:1000), Bax (1:1000), β-tubulin (1: 2000). After this, membranes were washed in TBST (0.05% Tween 20 (VWR International, Radnor, US), 0.025 mM Tris base, 0.1 M NaCl, pH 7.4) six times (5 min each) and incubated with HRP conjugated goat anti-mouse or goat anti-rabbit secondary antibodies (1:3000 in 5% non-fat milk in TBS) for 1 h at room temperature. Following incubation, membranes were washed with TBST six times (5 min each). Protein bands were visualised using SuperSignal West Pico Chemiluminescent Substrate and CL-XPosure film (Thermo Fisher Scientific, Waltham, Massachusetts, US). The developing process consisted of exposure to developer fluid, followed by a distilled water wash, then washed in fixer fluid Carestream Health (Rochester, New York, US) and lastly washed again in distilled water.
Densitometric analysis
The images were scanned using Bio-Rad GS-710 densitometer and densities of each band were quantified using Quantity One software (Bio-Rad). The densities of the respective proteins were normalised to β-tubulin. Three independent experiments were performed. Data is presented as mean ± SEM. Statistical significance was determined using one-way ANOVA with Bonferroni post hoc test using Prism 9 (GraphPad Software, US).
Cell viability was normalised to control and analysed by nonlinear regression using GraphPad Prism 9 software. Drug concentrations were log-transformed using the X = log(X) function. Nonlinear regression analysis was carried out using the log (inhibitor) versus response – four parameter variable slope function. Time course cell viability assays were plotted as cell number versus spiroleucettadine concentration and analysed with nonlinear regression using GraphPad Prism 9 software. This provided EC50 values in different cell lines and at different drug exposure lengths. Comparison of mean spiroleucettadine EC50 values in different cell lines was done using an extra sum of squares F test. Identical statistical analysis was used to compare spiroleucettadine EC50 values following differing cell exposure times to spiroleucettadine. Data is presented as mean ± SD, where p < 0.05 was the minimum requirement for statistically significant difference. Experiments were carried out in triplicate using three biological replicates.
Cell cycle and apoptosis data was gathered using FlowJo 10 software. Comparison between treatments was carried out using GraphPad Prism 9 software. To determine whether different concentrations of spiroleucettadine caused changes to cell numbers in each stage of the cell cycle, or to determine whether cells were viable or apoptotic, data was analysed using two-way ANOVA coupled with post hoc Bonferroni tests. Data is presented as mean ± SEM, where p < 0.05 was the minimum requirement for statistically significant difference. Experiments were carried out in triplicate using three biological replicates.
Statistical analysis of Western blot data was carried out by calculating the relative expression of the protein of interest in comparison to the β-tubulin loading control. To assess whether various concentrations of spiroleucettadine caused changes in protein expression, one-way ANOVA were used coupled with post hoc Bonferroni tests. Data are presented as mean ± SEM where p < 0.05 was the minimum requirement for statistically significant difference. Experiments were carried out using three biological replicates.
We first confirmed the sensitivity of human adenocarcinoma cell lines (H522, A549, and H1322) using the SRB assay to determine cell viability at various concentrations of spiroleucettadine. The EC50 concentration of spiroleucettadine with respect to cell viability was 0.72 ± 0.09 μM in H522 cells, the lowest of the three cell lines tested (Table 1).
Data are expressed as mean plus or minus standard deviation and p values were calculated from 1-factor ANOVA with cell type as the independent variable.
| A | H522 cells | A549 cells | H3122 cells | P-value |
|---|---|---|---|---|
| EC50 (μM) for cell viability | 0.72 ± 0.05 | 3.60 ± 0.02 | 1.59 ± 0.06 | <0.001 |
| B | H522 cells | |||
|---|---|---|---|---|
| EC50 (μM) for cell viability | 3 Day Treatment Exposure | 4 Day Treatment Exposure | 6 Day Treatment Exposure | P-value |
| EC50 (μM) | 0.66 ± 0.17 | 0.09 ± 0.16 | 0.09 ± 0.16 | 0.874 |
To test whether the EC50 for H522 cells sensitivity to spiroleucettadine varies with the amount of time the cells are exposed to spiroleucettadine we measured H522 cell number across a range of spiroleucettadine concentrations when spiroleucettadine was applied to cells for three, four or six days (this controls for the changing numbers of cells in the control wells, to which drug treated cells are normalised in the method used for Figure 1A). This showed that duration of drug exposure had no significant effect on cell number (Figure 1B) confirming the relative sensitivity of H522 cells to spiroleucettadine compared to A549 and H3122 cells (Table 1B). We therefore used H522 cells and spiroleucettadine concentrations calculated from 0.72 μM (the 3-day EC50 for cell viability) in the experiments described below. Furthermore, from the high degree of cell growth suppression at three days following exposure to this drug concentration, we inferred that significant cell cycle, apoptosis, and protein expression changes, would be detectable after 24 hours (when cellular processes have been activated, but cell number are sufficient for measurements to be robust).
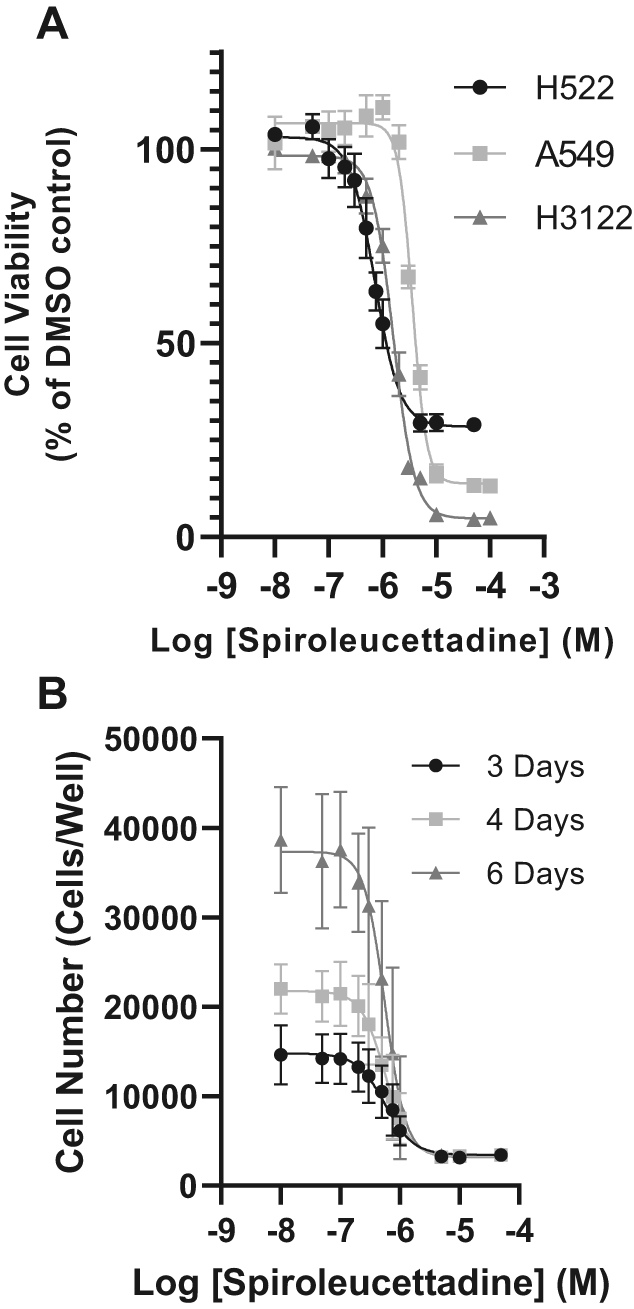
(A) Effect of a range of concentrations of spiroleucettadine on the viability of H522 (circles), H3122 (triangles), and A549 cells (squares). Data points are displayed as mean ± SD (n = 3 biological replicates). (B) Effect of duration of exposure to various concentrations of spiroleucettadine on H522. Cell number was counted after 3, 4 or 6 days exposure to spiroleucettadine. Data are displayed as mean ± SEM (n = 3 biological replicates).
To characterise the mode of cell growth suppression by spiroleucettadine more precisely, we assayed its effects first on the cell cycle and then on cell cycle mediators. No significant changes in cell number were detected in the sub G1, G0/G1, S and G2/M phases following 24-hour treatment with spiroleucettadine at 0.72 μM (1×EC50) or 0.36 μM (0.5×EC50) (Figure 2A). However, 1.44 μM (2×EC50) resulted in a significant decrease (~15%) in cells in the G0/G1 phase (p < 0.001) and a significant increase (~35%) in cells in the G2/M phase (p < 0.0001) (Figure 2A) suggesting that spiroleucettadine increases the number of cells in G2/M arrest.
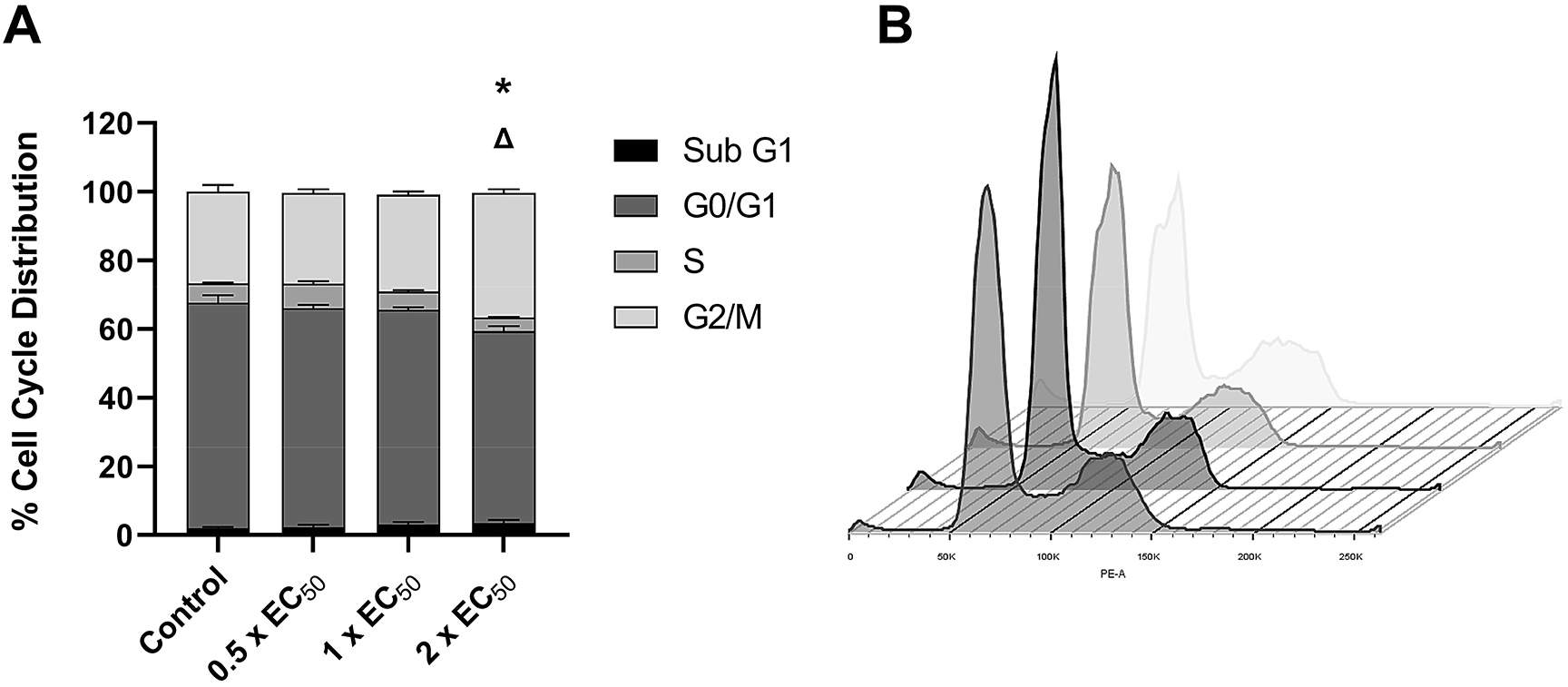
(A) Effect of spiroleucettadine on cell cycle arrest in H522 cells following 24 hours exposure to vehicle control (DMSO 0.1%) or 0.5 × cell viability assay EC50 (0.36 mM), 1 × EC50 (0.72 mM) and 2 × EC50 (1.44 mM) of spiroleucettadine. Bar denotes;  Sub G1,
Sub G1,  G0/G1,
G0/G1,  S,
S,  G2/M. (B) Representative flow cytometry cell cycle histogram. Differently shaded histograms denote different treatments (from front to back: control; 0.36 mM; 0.72 mM; 1.44 mM). Data analysed with two-way-ANOVA followed by Bonferroni post-hoc tests and are presented as mean ± SEM (n = 3 biological replicates). * p < 0.05 of cells in G0/G1 phase compared with control; Δ p < 0.05 of cells in G2/M phase versus all control.
G2/M. (B) Representative flow cytometry cell cycle histogram. Differently shaded histograms denote different treatments (from front to back: control; 0.36 mM; 0.72 mM; 1.44 mM). Data analysed with two-way-ANOVA followed by Bonferroni post-hoc tests and are presented as mean ± SEM (n = 3 biological replicates). * p < 0.05 of cells in G0/G1 phase compared with control; Δ p < 0.05 of cells in G2/M phase versus all control.
We did not detect any statistically significant changes in protein expression using Western blotting for CDK1, CDK4 or the phosphorylation of CDK4 (pCDK4) after 24-hour treatment with spiroleucettadine at concentrations up to 1.4 μM (2 × EC50) (p > 0.05) (Figure 3A, C-E). A significant increase (~50% and ~95%, respectively) in the expression of pCDK1, was, however, observed following treatment with 0.72 μM and 1.44 μM spiroleucettadine for 24 hours (p < 0.01) (Figure 3B).

Treatment conventions are as above. for 24 hours. Representative Western blots and densitometry (n = 3 biological replicates) are shown for: (A) CDK1; (B) pCDK1; (C) CDK4; (D) pCDK4. Data are expressed as mean ± SEM determined and analysed by one-way-ANOVA with Bonferroni post-hoc tests (n = 3 biological replicates). * p < 0.05.
When we assayed for changes in expression of cyclin B1 and cyclin D1 in H522 cells following 24-hour exposure to spiroleucettadine using Western blotting, we found a significant increase in cyclin B1 expression in response to both 0.72 μM and 1.44 μM spiroleucettadine for 24 hours (p < 0.05) (~45% higher than that of the control treatment group) (Figure 4A). Cyclin D1 expression was significantly reduced (p < 0.05) by all concentrations of spiroleucettadine tested: ~35% for 0.36 μM (0.5 × cell viability EC50); ~38% for 0.72 μM (EC50); and ~60% for 1.42 μM (2 × EC50) (Figure 4B).
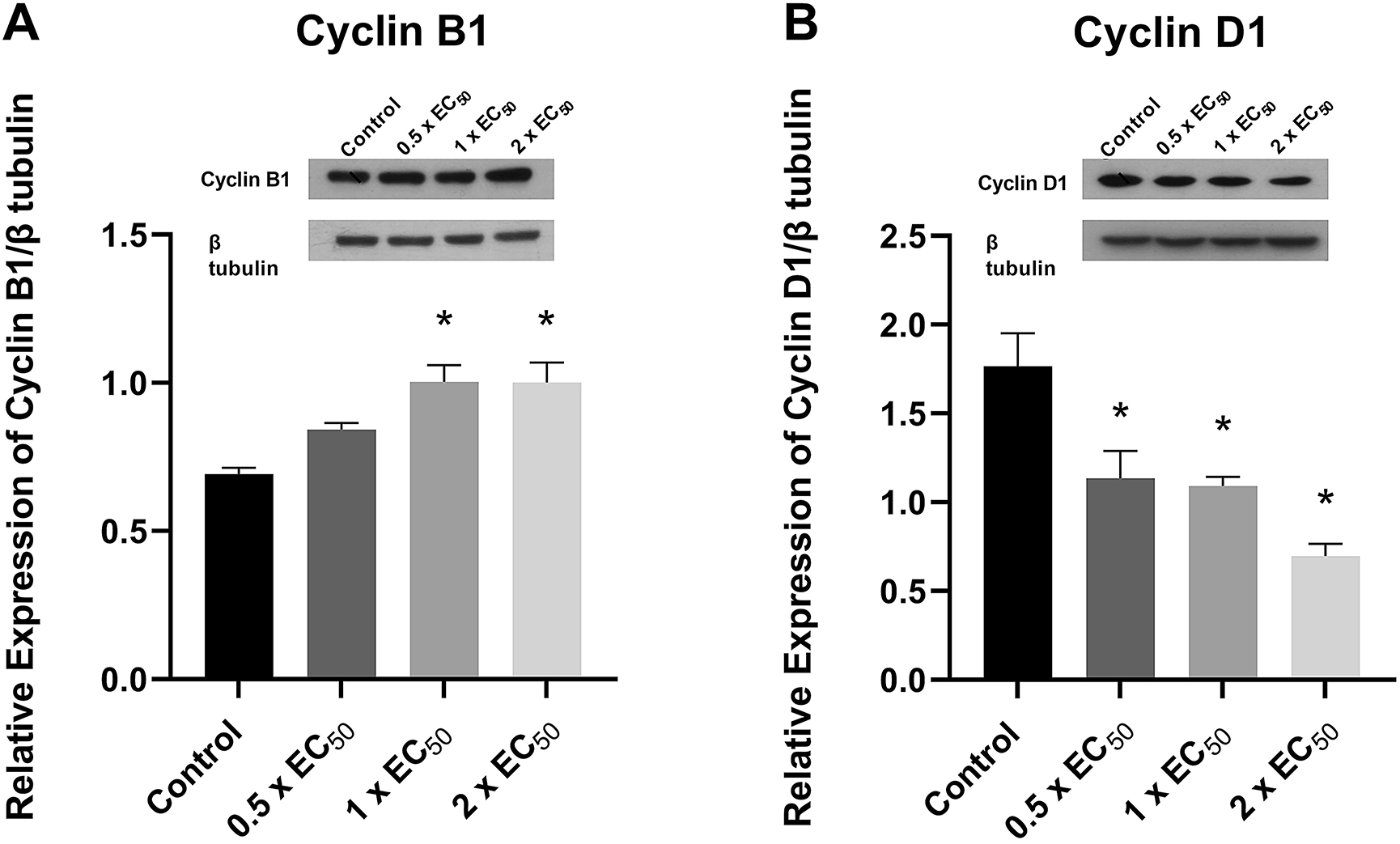
Treatment and densitometry conventions are as above. Representative Western blots and densitometry (n = 3 biological replicates) are shown for: (A) cyclin B1; (B) cyclin D1.
To characterise the induction of apoptosis in H522 cells by spiroleucettadine we used annexin V and propidium iodide staining with flow cytometry. A significant increase in the percentage of cells in the early apoptotic stage was observed following 1.42 μM (2 × cell viability EC50) treatment when compared control (p < 0.05) (Figure 5). The increase, while statistically significant, was relatively small with ~0.5% cells in the early apoptotic phase in the control treatment group compared to ~2.3% in spiroleucettadine-treated cells. In addition, 1.42 μM spiroleucettadine over 24 hours produced a significant (p < 0.001) increase in the percentage of cells in the late apoptotic phase in comparison to all lower concentration treatments tested (Figure 5). The percentage of cells in the late apoptotic phase ranged from ~2-3% in the control cells to ~11% in treated cells (Figure 5).
 Early apoptotic, late apoptotic. Data were analysed with two-way-ANOVA followed by Bonferroni post-hoc tests and are presented as mean ± SEM (n = 3 biological replicates). * p < 0.0001 late apoptosis versus control, Δ p < 0.05 early apoptosis versus control. (B-E) Representative flow cytometry plots of Annexin V staining following 48 hours exposure to spiroleucettadine (from left to right: control; 0.36 mM; 0.72 mM; 1.44 mM).
Early apoptotic, late apoptotic. Data were analysed with two-way-ANOVA followed by Bonferroni post-hoc tests and are presented as mean ± SEM (n = 3 biological replicates). * p < 0.0001 late apoptosis versus control, Δ p < 0.05 early apoptosis versus control. (B-E) Representative flow cytometry plots of Annexin V staining following 48 hours exposure to spiroleucettadine (from left to right: control; 0.36 mM; 0.72 mM; 1.44 mM).To characterise the drivers of apoptosis induced by spiroleucettadine more precisely we assayed changes in protein expression of apoptosis mediators Bax, Bcl-2, Bim, and cleaved caspase 3 in H522 cells. After 24 hours of treatment, the expression of neither Bax nor Bcl-2 was significantly changed by any concentrations of spiroleucettadine tested (p > 0.05, Figure 6). The expression of Bim was substantially and significantly increased following treatment by both 0.72 μM and 1.44 μM spiroleucettadine over 24 hours (~83% and ~150%, respectively, p < 0.01) (Figure 6). Cleaved caspase 3 expression was substantially and significantly increased (~385%) following treatment with 1.44 μM spiroleucettadine over 24 hours (p < 0.0001) (Figure 6).
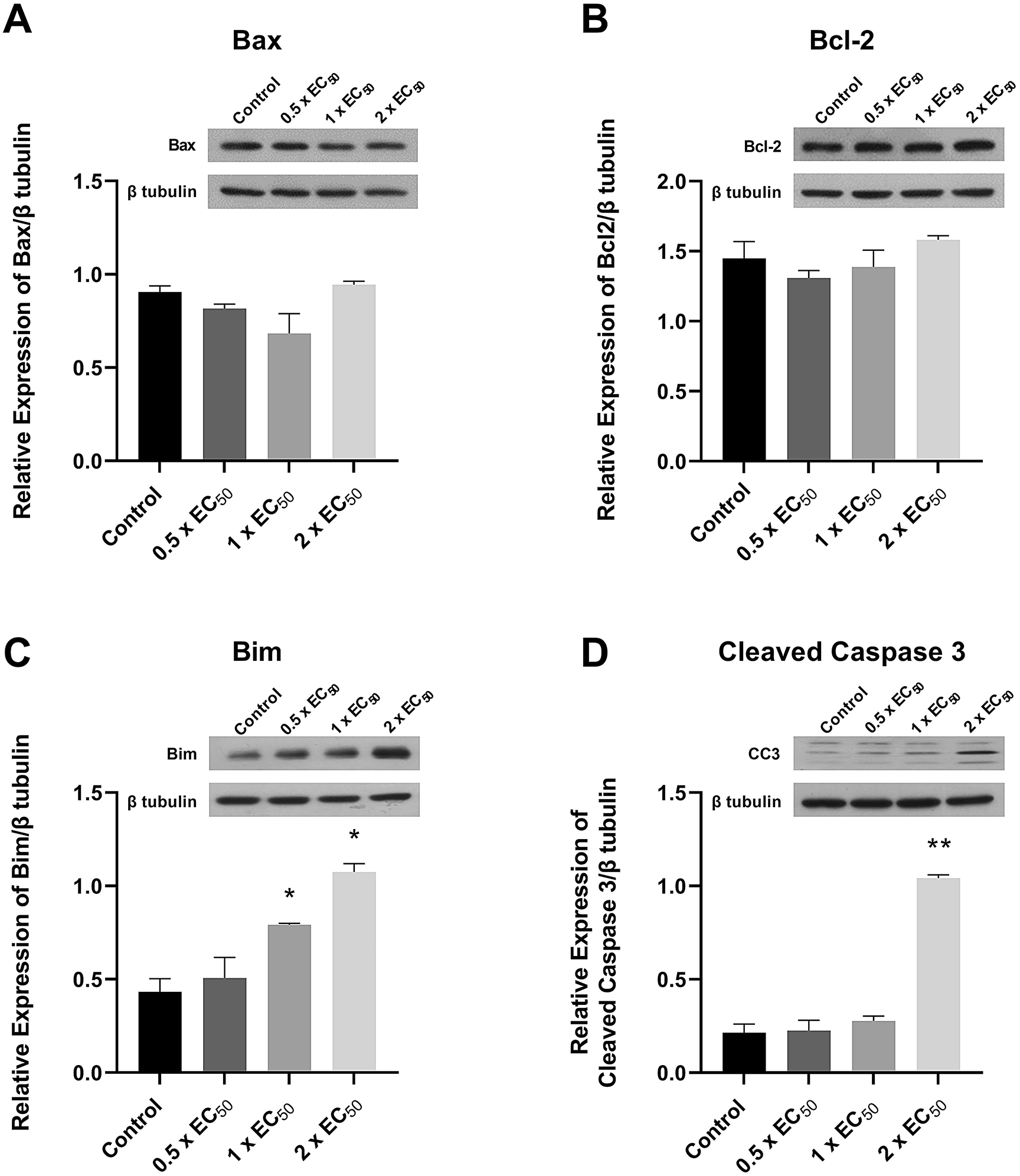
* p < 0.01, ** p < 0.0001 versus control. Treatment and densitometry conventions are otherwise as above. Representative Western blots and densitometry (n = 3 biological replicates) are shown for: (A) Bax; (B) Bcl-2; (C) Bim; (D) cleaved caspase 3.
The identification of new anticancer agents for non-oncogene dominant NSCLC has been challenging. Although considerable progress has been made in the treatment for EGFR, ALK, and PDL-1 positive subtypes, NSCLC is generally resistant to chemotherapy. Building on recent work showing potential anti-cancer properties of spiroleucettadine (Badart, Barnes et al., 2020), we first identified a cell line (H522) with particular sensitivity to the compound and then characterised the mode and mediators of cell growth suppression.
We found that in lung adenocarcinoma H522 cells, spiroleucettadine at 1.44 μM decreases the number cells in the G0/G1 phase of the cell cycle and increased the proportion of cells in the G2/M phase over 24 hours, suggesting a G2/M arrest. Consistent with this, we found a strong increase in the phosphorylation of CDK1 and expression of cyclin B1 and cyclin D1 over the same period, both mediators of cell cycle transition from G2/M. Cyclin D1 expression – a mediator of the G0/G1 transition expression – was significantly reduced. Apoptosis was also induced by spiroleucettadine, evidenced by both flow cytometry and increased expression of cleaved caspase 3. Expression of Bim, a mediator of the intrinsic apoptosis pathway, was profoundly induced by spiroleucettadine. The picture that emerges from these results is a familiar one from induction of apoptosis by cell cycle arrest and activation of mitotic checkpoints by cytotoxic chemotherapies.
We hypothesised that by analogy with other natural product derivatives, namely taxanes, vincra alkaloids, and in particular eribulin, the mechanism of action of spiroleucettadine might also be disrupting microtubules in mitotic spindle formation, which would be consistent with the results discussed above, namely G2/M arrest, as cells in the G2 phase are preparing for mitotic division in the M phase including mitotic spindle formation (Schmidt, Rohe et al., 2017). However, although this hypothesis deserves more extensive investigation, we did not find any evidence of this using immunocytochemistry for α-tubulin, which has previously been shown for microtubule disrupters (Florian and Mitchison, 2016).
Our experiments did not show any evidence of an induction of cell cycle arrest at G1 which aligns with the absence of p53 in H522 cells; cells without p53 cannot be arrested in G1 (Murray, 1994). This raises the question of why H522 cells were more sensitive to cell growth suppression than other cells tested. It may be that H522 cells lack a tumour suppressor, p53, but not a positive driver of cell growth, such as ALK, EGFR, or KRAS as for the other cell lines tested. This may make H522 cells more sensitive to drugs that disrupt the cell cycle than the other cell lines. However, loss of tumour suppression mediators is generally associated with resistance anticancer therapeutics, not sensitivity (Song and Xu, 2007, Goldstein, Marcel et al., 2011).
The increase of phosphorylation of CDK1 by spiroleucettadine may offer another avenue for further investigations into mechanism of action. Phosphorylation of WEE1 and Myt1 kinases on tyrosine 15 (Tyr15) lead to the phosphorylation of CDK1. Also, CDK-activating kinases (CAKs) phosphorylate CDK1 at threonine 161 (T161) (Atherton-Fessler, Parker et al., 1993, Pavletich 1999). In addition, the induction of Bim – a Bcl-2 homology 3 (BH3) only pro-apoptotic – implies activation of the intrinsic apoptosis pathway (Youle and Strasser, 2008, Tzifi, Economopoulou et al., 2012, Warren, Wong-Brown et al., 2019).
Spiroleucettadine has sub-micromolar suppression of H522 cancer cell growth, through G2/M arrest and induction of the intrinsic apoptosis pathway. This is associated with the phosphorylation of CDK1 and the expression of Bim, respectively. Proposed targets for spiroleucettadine are thereby constrained to those with mechanisms of actions that lead to these cellular and biochemical changes.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)