Keywords
bacterial nomenclature, bacterial taxonomy, concatenated phylogeny, species-specific barcode reference library
This article is included in the Manipal Academy of Higher Education gateway.
This article is included in the Cell & Molecular Biology gateway.
bacterial nomenclature, bacterial taxonomy, concatenated phylogeny, species-specific barcode reference library
The 16S ribosomal RNA (16S rRNA) encoding region is extensively studied to identify and classify bacterial species. The 16S rRNA is a conserved component of the 30S small subunit of a prokaryotic ribosome. The gene is ~1500 base pair (bp) long, and it consists of nine variable regions (Reller et al. 2007; Sabat et al. 2017). For decades, the sequence of the 16S rRNA gene has been used as a potential molecular marker in culture-independent methods to identify and classify diverse bacterial communities (Clarridge, 2004; Johnson et al. 2019). The 16S rRNA sequences are currently being used as an accurate and rapid method to study bacterial evolution, phylogenetic relationships, populations in an environment, and quantification of abundant taxa (Vetrovsky and Baldrian, 2013; Srinivasan et al. 2015; Peker et al. 2019).
Despite the wide range of applications, a few shortcomings limit the accuracy of results derived through the 16S rRNA sequence analysis. One such aspect is that the 16S rRNA gene has poor discriminatory power at the species level (Winand et al. 2020), and the copy number can vary from 1 to 15 or even more (Vetrovsky and Baldrian, 2013; Winand et al. 2020). The presence of multiple variable copies of this gene makes distinct data for a species. Hence, gene copy normalization (GCN) is necessary prior to sequence analysis. However, studies show that the GCN approach does not improve the 16S rRNA sequence analyses in real scenarios and suggests a comprehensive species-specific catalogue of gene copies (Starke et al. 2021). Secondly, the intra-genomic variations between the 16S rRNA gene copies were observed in several bacterial genome assemblies (Paul et al. 2019). Only a minority of the bacterial genomes harbor identical 16S rRNA gene copies, and sequence diversity increases with increasing copy numbers (Vetrovsky and Baldrian, 2013). Further, currently available 16S rRNA-based bioinformatics approaches are not always amenable to classify bacterium at the species level due to high inter-species sequence similarities (Peker et al. 2019; Deurenberg et al. 2017).
A few other issues are also related to the sequencing and bioinformatics analysis of 16S rRNA gene regions. These include the purity of bacterial isolates, the quality of isolated DNA, and the possibility of chimeric molecules (Janda and Abbott, 2007; Church et al. 2020). Base-call errors can also mislead the sequence identity and phylogenetic inferences (Alachiotis et al. 2013). The other concerns on sequence-based analysis, comparison, and species identification include the number of base ambiguities processed, gaps generated during sequence comparison, and algorithm (local or global) used for the sequence alignment. The local alignment algorithm is extensively used for sequence similarity-based species identification. Several studies were conducted to identify the best variable region or combination of variable regions for bacterial classification, and a consensus remains to be implemented (Janda and Abbott, 2007; Johnson et al. 2019; Winand et al. 2020). Usage of misclassified sequence as a reference and improper bioinformatics workflows mislead the bacterial taxonomy. Further, the growth of bioinformatics and genetic data has placed genome-based microbial classification with researchers with little or no taxonomic experience, which may also mislead the bacterial taxonomy (Baltrus, 2016).
A few bacterial identification systems with high resolution have been developed using the sequence of polymerase chain reaction (PCR) amplified ∼4.5 kb long 16S–23S rRNA regions (Benítez-Páez and Sanz, 2017; Sabat et al. 2017; Kerkhof et al. 2017). However, these approaches have a few limitations, such as the lack of reference 16S–23S rRNA sequence databases and complementary bioinformatics resources for reliable species identification (Sabat et al. 2017). The recent advancements in bioinformatics workflows (Winand et al. 2020; Schloss, 2020) and reference databases such as SILVA, EzBioCloud (Quast et al. 2013; Yoon, 2017) improved 16S rRNA-based bacterial taxonomy. However, a few recent genome-based studies highlighted the misclassification incidences in bacterial species and genome assemblies (Steven et al. 2017; Martínez-Romero, et al. 2018; Mateo-Estrada et al. 2019; Bagheri et al. 2020).
Nowadays, conventional and high throughput sequencers can amplify all the nine variable regions of the 16S rRNA gene. Although, many 16S rRNA-based bacterial identification studies lack a complete set of variable regions (Stackebrandt et al. 2021). The classical and high throughput sequencing technologies produce a large volume of whole-genome data. There is an urgent need to translate the genomic data for convenient microbiome analyses that ensure clinical practitioners can readily understand and quickly implement it (Church et al. 2020). Hence, the study intended to demonstrate a workflow to develop species-specific concatenated 16S rRNA reference libraries and its analysis. The species-specific libraries can yield better resolution in sequence similarity and phylogeny based bacterial classification approaches.
Sequence alignment of 16S rRNA copies at the intra-genomic level shows a higher degree of variability in species belonging to the Firmicutes and Proteobacteria (Vetrovsky and Baldrian, 2013; Ibal et al. 2019). Hence, the study used eight 16S rRNA copies (Underlying data: Supplementary data 1 (Paul, 2022)) retrieved from the whole genome of Enterobacter asburiae strain ATCC 35953 (NZ_CP011863.1). The BLAST+ 2.13.0 (RRID:SCR_004870; Altschul et al. 1990) and Clustal Omega 1.2.4 (RRID:SCR_001591; Sievers et al. 2011) sequence alignment algorithms were used to estimate intra-genomic variability between the 16S rRNA gene copies. Phylogenetic relatedness between intra-genomic 16S rRNA copies were estimated using the Maximum Likelihood method (Tamura-Nei model; 500 bootstrap replicates) with MEGA software (version 11; RRID: SCR_000667; Kumar et al. 2018).
Previous studies have reported that several bacterial species share more than 99% sequence identity in the 16S rRNA encoding region. Hence, the 16S rRNA-based bacterial identification methods failed to discriminate such genetically related species (Deurenberg et al. 2017; Devanga-Ragupathi et al. 2018). It has been reported that Streptococcus mitis and Streptococcus pneumoniae are almost indistinguishable from each other based on the sequence similarity of their 16S rRNA regions (Reller et al. 2007; Lal et al. 2011). To develop species-specific barcode reference libraries, the study used 16S rRNA gene copies from whole-genome assemblies of four closely related species of Streptococcus (S. gordonii, S. mitis, S. oralis, and S. pneumoniae).
More than 463,000 whole-genome assemblies are currently available for prokaryotes at the Genome database (RRID:SCR_002474; https://www.ncbi.nlm.nih.gov/genome). Most microbial genomes were sequenced with high throughput sequencing technologies such as Illumina/Ion-Torrent (short read sequencing) and PacBio/Nanopre (long read sequencing). Further, many of these whole-genome assemblies are derived through a hybrid assembly of short and long read sequence data. The large volume of high throughput data can be effectively used to develop advanced genome-based approaches for microbial systematics. The genomic data is available in four assembly completion levels (contig, scaffold, chromosome, and complete). However, the study used only the genomes assemblies in the 'complete' stage to retrieve 16S rRNA gene copies.
The study retrieved full-length 16S rRNA gene copies from 16 genome assemblies belonging to four Streptococcus species (S. gordonii, S. mitis, S. oralis, and S. pneumoniae). The detailed information on the dataset used to develop species-specific concatenated reference libraries is provided in Table 1 and the sequences are provided in the underlying data (Supplementary data 2 (Paul, 2022)). To maintain equal length, sequences were trimmed out beyond the universal primer pair fD1-5'-GAG TTT GAT CCT GGC TCA-3' and rP2-5'-ACG GCT AAC TTG TTA CGA CT-3' (Weisburg et al. 1991) for full-length 16S rDNA amplification. The study used MEGA 11 software to perform multiple sequence alignment and identify the intra-species parsimony informative (Parsim-info) variable sites. A species-specific barcode reference library covering entire Parsim-info variable sites was constructed by concatenating four 16S rRNA gene copies representing four different strains of a species. The rationale behind the selection of four copies for a species-specific barcode reference library is: (i) a maximum of four variations can be found on a single site, and (ii) earlier studies have shown that the mean 16S rRNA copies per genome is four (Vetrovsky and Baldrian, 2013).
The study analyzed a few cases to demonstrate the classical sequence similarity and phylogenetic analysis using concatenated species-specific 16S rRNA reference libraries. The study used nine Sanger sequenced 16S rRNA gene copies showing higher sequence similarity with multiple species of Streptococcus retrieved from the GenBank database (RRID:SCR_002760). The web based BLAST2 (version 2.13.0) program for aligning two or more sequences was used to estimate the maximum score, total alignment score, and sequence identity. A single copy of the 16S rRNA region derived through Sanger sequencing or retrieved from a whole-genome assembly can be considered as ‘Query sequence’. The concatenated species-specific reference libraries must be provided in the ‘Subject sequence’ section. To perform phylogenetic analysis, it is mandatory that the target sequence (length = n bp) has to be concatenated four times (length = 4 × n bp), appending next to the last base. Phylogenetic relatedness was estimated using the Maximum Likelihood method (Tamura-Nei model; 500 bootstrap replicates) with MEGA 11 software.
Historically, the 16S rRNA gene sequences were used to identify known and new bacterial species. However, this method is impacted by several factors such as amplification efficiency, poor discriminatory power at the species level, multiple polymorphic 16S rRNA gene copies, and improper bioinformatics workflows for the data analysis. The E. asburiae genome had eight 16S rRNA gene copies that showed a mean identity of 99.29% in sequence alignment using Clustal Omega (global alignment), whereas BLAST (local alignment) analysis resulted in an average of 99% identity between the copies (Table 2). Hence, the selection of an appropriate algorithm has a significant role in the estimation of percent identity, and a vital role in sequence-based species delineation. Global sequence alignment programs generally perform better for highly identical sequence pairs, and the algorithm considers all the bases for the estimation of sequence identity. The multiple sequence alignment showed 22 variable sites in 16S rRNA gene copies of the E. asburiae genome (Figure 1).
Percent identity given below the diagonal line is calculated with Clustal Omega software (Mean identity: 99.29%) and those above the diagonal line were calculated with the BLASTN program (Mean identity: 99.00%). Genome coordinates of 16S rRNA copies: R1: 2686082–2687660 (1579 bp); R2: 3148265–3149814 (1550 bp); R3: 3313470–3315019 (1550 bp); R4: 3583942–3585481 (1540 bp); R5:3684745–3686294 (1550 bp); R6: 3771751–3773300 (1550 bp); R7: 3968538–3970087 (1550 bp); R8: 4647650–4649199 (1550 bp)
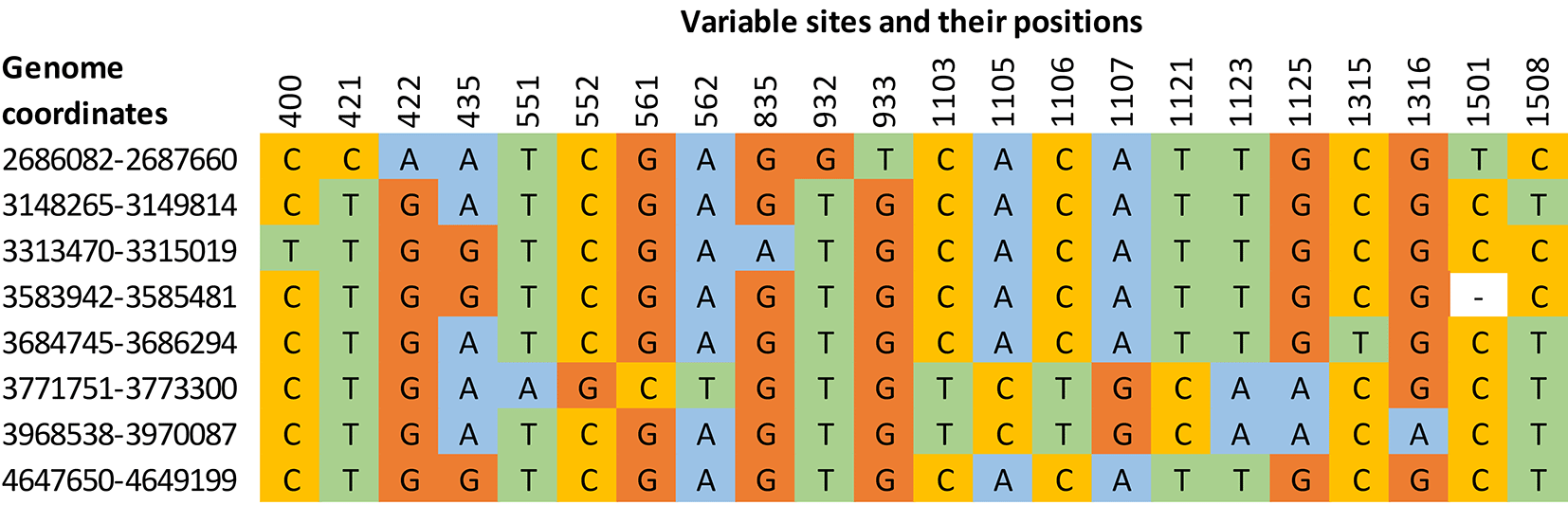
The evolutionary relationship between species is usually represented in a phylogenetic tree drawn using a single barcode gene, multiple genes, or whole genomes. However, bacterial species nomenclature is mainly designated based on the confidence obtained from the phylogenetic tree derived through single copy 16S rRNA analysis. To highlight how the intra-genomic 16S rRNA variations influence the species delineation, a phylogenetic tree was constructed using eight 16S rRNA gene copies of E. asburiae reference genome showing multiple nodes (Figure 2). The sequence similarity and phylogeny-based analysis indicate that the intra-genomic variations in 16S rRNA copies may mislead the bacterial taxonomy in single gene copy approaches.
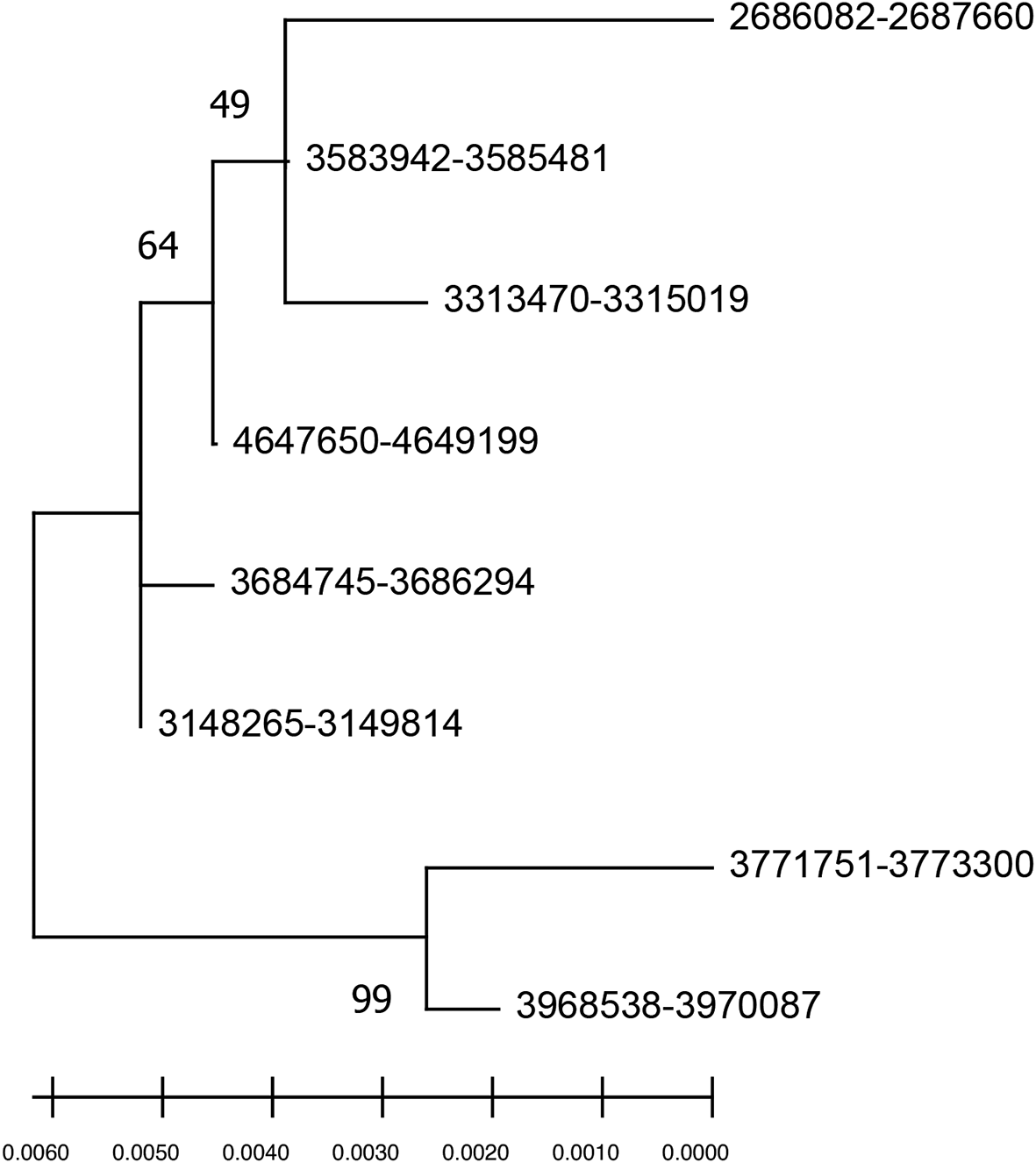
The node label denotes the coordinate of 16S rRNA regions in the genome.
The study selected four Streptococcus species (S. gordonii, S. mitis, S. oralis, and S. pneumoniae) to construct species-specific concatenated 16S rRNA reference libraries. The study used 16S rRNA copies retrieved from four whole genome assemblies in the ‘complete’ stage to construct a species-specific barcode library. Four copies of the 16S rRNA gene are required to construct the concatenated library for a species. The details of constructed species-specific libraries are listed in Table 1 and the sequence is provided in the underlying data (Supplementary data 3 (Paul, 2022)). The 16S rRNA sequence analysis shows 24 Parsim-info variable sites for S. oralis, 11 variations in S. mitis, seven variations in S. gordonii, and six variations found in S. pneumoniae. The observed intra-species Parsim-info variable sites are residing on both conserved and variable regions of the 16S rRNA gene.
The study used full-length 16S rRNA copies from four different strains to highlight the variations at the species level. However, a large volume of partial 16S rRNA sequences are available in the public genetic databases. In such cases, a species-specific concatenated 16S rRNA reference library can be developed with partial sequences. Intra-species variation on 16S rRNA gene copies influences the sequence based bacterial taxonomy. Hence, the concatenated 16S rRNA approach yields better resolution than single copy analysis in classical sequence similarity and phylogeny based species identification approaches.
The study compared nine 16S rRNA sequences representing Streptococcus species (Table 3) with species-specific concatenated reference libraries. Concatenated sequence analysis gives better resolution in sequence similarity search and phylogenetic analysis. The sequence accession numbers GU470907.1 and KF933785.1 classified as S. mitis showed a higher maximum and total alignment score with S. oralis than S. mitis (Table 3). Whereas the sequence (OM368574.1; classified as S. mitis) showed a higher sequence alignment score with S. pneumoniae. Figure 3A shows a maximum likelihood tree of the nine 16S rRNA gene sequences with four concatenated species-specific reference libraries. The concatenated GU470907.1 and KF933785.1 sequences showed a phylogenetic relationship with S. oralis and sequence OM368574.1 was genetically related to S. pneumoniae. These results indicate that the species-specific concatenated 16S rRNA reference libraries have great potential in the taxonomic classification. Hence, the study suggests the usage of concatenated variable 16S rRNA copies for sequence similarity and phylogeny-based species identification. A species-specific reference library with concatenated 16S rRNA gene copies provides better resolution in phylogenetic analysis than the single copy inference.
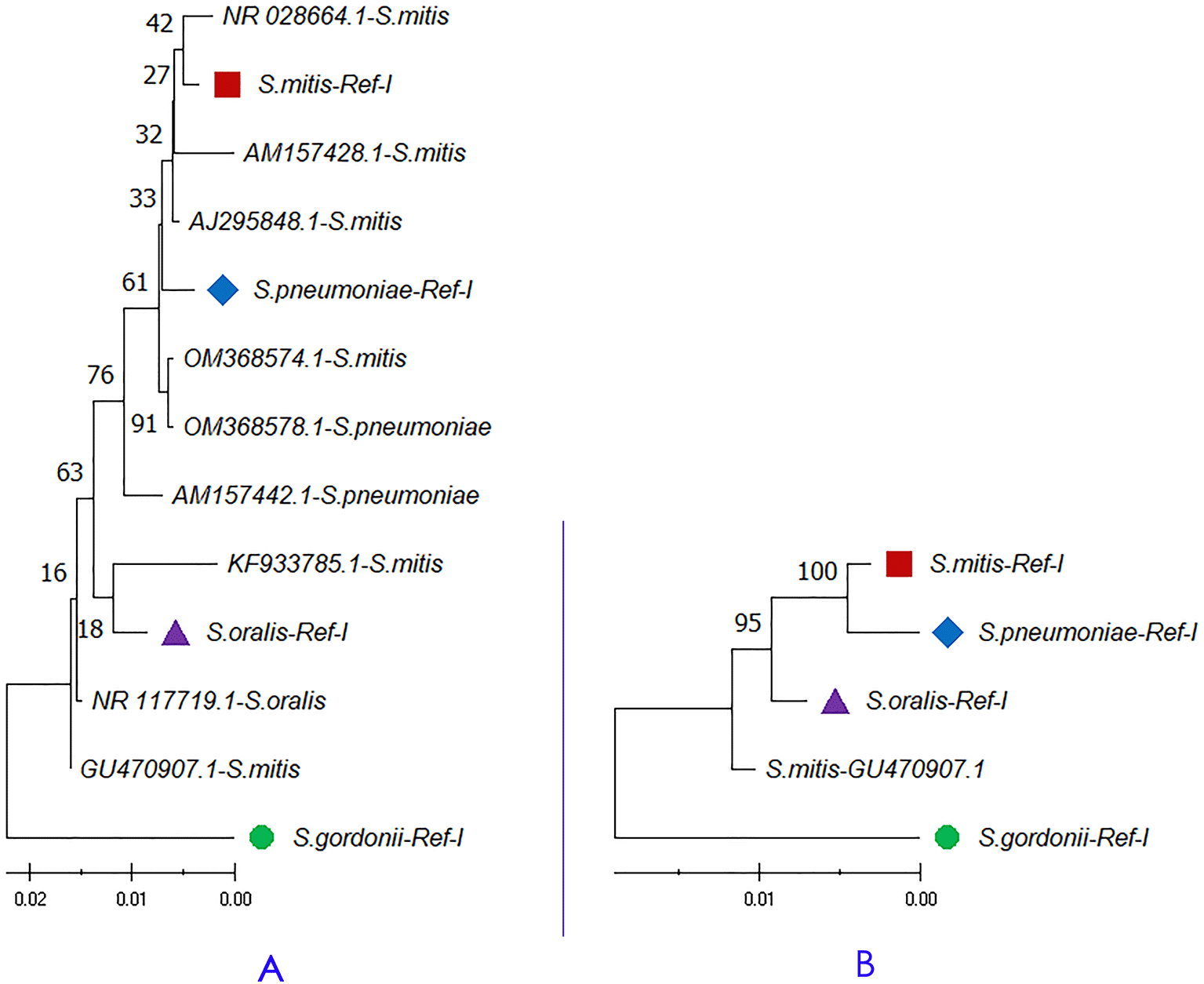
A) Phylogenetic analysis of randomly selected 16S rRNA sequences classified as Streptococcus species.
B) Concatenated 16S rRNA phylogeny of Streptococcus mitis sequence (Accession Number GU470907.1) showed 100% identity with Streptococcus oralis genome (Accession Number CP034442.1) in a BLAST based sequence similarity search. The node name highlighted in shapes (●, ■, ▲, ◆) represents the four species-specific reference libraries.
Sequencing and analysis of the 16S rRNA encoding region is considered a conventional and robust method for identifying and classifying the bacterial species. The barcode gene is widely used in sequence similarity, phylogeny, and metagenome-based species identification. However, the accuracy of bacterial taxonomy based on 16S rRNA barcode regions is limited by the intra-genomic heterogeneity of multiple 16S rRNA gene copies and significant sequence identity of this gene between closely related taxa. Further, discrimination of closely related species identification through sequences of the 16S rRNA gene is a challenge, and it may lead to species misidentification (Boudewijns et al. 2006; Church et al. 2020). About 15% of the genomes have only a single copy of the 16S rRNA gene, and only a minority of bacterial genomes harbour identical 16S rRNA gene copies (Vetrovsky and Baldrian, 2013). The 16S rRNA gene copies can vary from 1 to 15 in a genome, and the copy number of variations is taxon specific (Vetrovsky and Baldrian, 2013). Sequence diversity increases with the increasing 16S rRNA copy numbers. The 16S rRNA sequence variation can even be found at intra-genomic level or in different strains of a species as well. Amplification of limited number of variable regions cannot achieve the taxonomic resolution achieved by sequencing the entire gene (Johnson et al. 2019). Usage of misclassified 16S rRNA sequences as a reference and inappropriate bioinformatics workflows also mislead the taxonomic assignment. To overcome these challenges, it is important to translate high throughput microbial genomic data into meaningful, actionable information that clinicians can readily understand and quickly implement for bacterial identification. Hence, the study intended to develop a species-specific catalogue of concatenated 16S rRNA gene copies that can yield better inference in sequence similarity and phylogenetic analysis.
Several bioinformatics resources are extensively used for the 16S rRNA sequence analysis and bacterial identification. However, several researchers report the sequence similarity derived through a local alignment algorithm. Earlier reports have suggested that the species belonging to the taxa Gammaproteobacteria show higher intra-species variability (Vetrovsky and Baldrian, 2013). Hence, the study estimated the percent identity of intra-genomic 16S rRNA gene copies of Enterobacter asburiae using local and global alignment algorithms. The reference genome of E. asburiae has eight 16S rRNA gene copies in its genome. The BLAST and Clustal sequence alignment algorithms yielded marginally varying results for the intra-genomic 16S rRNA gene copies. Local alignment algorithms may not consider base mismatches at the sequence ends for calculating percent identity, while global alignment algorithms consider entire bases. Therefore, global sequence alignment is best for estimating intra and inter-species identity for single gene copies. However, BLAST can calculate the total alignment score with multiple paralogue regions. Hence, web-based BLAST2 is suggested for estimating the sequence similarity using concatenated barcode reference libraries.
The GenBank (Leray et al. 2019) and NCBI 16S RefSeq database for bacteria (Winand et al. 2020) are reliable for species-level identification and classification. However, few earlier studies have highlighted the misclassification of species and genome assemblies in public genetic databases (Parks et al. 2018; Varghese et al. 2015). For example, the 16S rRNA sequence accession number (Ac. No.) LT707617.1 shows the organism as Streptococcus mitis. Conventional BLAST-based sequence similarity search shows the highest identity of 99.60% with S. mitis 16S rRNA sequence (Ac. No. AB002520.1). However, the 16S rRNA sequence (Ac. No. LT707617.1) did not show significant similarity with other 16S rRNA reference sequences available for S. mitis. Further, the sequence also shows 99.44% identity with reference 16S rRNA sequences of S. gordonii. Hence, the study performed a sequence alignment of the sequence (Acc. No. LT707617.1) against species-specific concatenated 16S rRNA reference libraries for S. gordonii (S.gordonii-Ref-I), and S. mitis (S.mitis-Ref-I). The alignment resulted in a significant identity of 99.44% with S.gordonii-Ref-I (2279 maximum and 9041 total alignment score) than S.mitis-Ref-I (97.13% identity with 2119 maximum and 8449 total alignment score). Single copy BLAST results may show only a minor fraction of the difference in percent identity and maximum or total alignment score for closely related species. However, sequence similarity estimation using species-specific concatenated reference libraries shows marginal difference in total alignment score, as it is aligned against four copies. Hence, 16S rRNA analysis with a species-specific concatenated barcode reference library will give better accuracy for bacterial classification than approaches using a single copy.
Several 16S rRNA sequences show 100% identity with multiple species, which is the major challenge in sequence-based species identification. For example, the 16S rRNA sequence from S. mitis (Ac. No. GU470907.1; 1522 bp) shares 100% identity with the 16S rRNA gene from S. oralis strain ATCC 35037 genome (Ac. No. CP034442.1). Hence, the sequence (GU470907.1) aligned against the species-specific concatenated reference libraries for S. oralis (S.oralis-Ref-I), and S. mitis (S.mitis-Ref-I). The result showed 100% identity with S. oralis (2787 maximum and 10936 total alignment score), and 99.14% identity with S. mitis (2715 maximum and 10796 total alignment score). Further, a phylogenetic tree of GU470907.1 (1509 × 4 = 6036 bp) with reference libraries S.mitis-Ref-I, and S.oralis-Ref-I was plotted. The maximum likelihood-based phylogenetic tree showed that the S. mitis (GU470907.1) sequence is more closely related to S. oralis than S. mitis (Figure 3B). Concatenated 16S rRNA-based estimation of sequence similarity and a phylogenetic inference provides better resolution than single-gene approaches. These results show that concatenated 16S rRNA approach is very effective in discriminating even genetically related bacterial species. Further, other studies also highlighted that the phylogenetic tree inferred from vertically inherited protein sequence concatenation provided higher resolution than those obtained from a single copy (Ciccarelli et al. 2006; Thiergart et al. 2014).
Recent phylogenetic studies using concatenated multi-gene sequence data highlighted the importance of incorporating variation in gene histories, which will improve the traditional phylogenetic inferences (Devulder et al. 2005; Johnston et al. 2019). Further, one type of analysis should not be relied upon, instead, and to a certain extent, integrated bioinformatics approaches can avoid misclassification. As a cost-effective approach, the study combined substantial variations in 16S rRNA gene copies from a species to examine the performance of the single gene concatenation approach. Analyses using a concatenated 16S rRNA gene approach have some advantages: (i) the gene is present in all the bacterial species, (ii) the gene is weakly affected by horizontal gene transfer, (iii) the approach is very cost-effective, (iv) there is a large volume of reference genomic data available for several bacterial species, (v) it is effective in discriminating closely related bacterial species, (vi) the analyses can be performed in a computer with minimum configuration, and (vii) the analyses can be employed with available tools for sequence similarity and molecular phylogeny.
The concatenated 16S rRNA analyses drew the following suggestions:
• Full-length 16S rRNA gene amplification provides better accuracy than inference from a partial gene with a limited number of variable regions.
• Prior to the analysis, trim the bases beyond the primer ends and correct the base-call errors, which will avoid several mismatches in the sequence alignment.
• Estimation of mean 16S rRNA identity at the intra-species level helps to classify the species having a higher degree of intra-genomic 16S rRNA heterogeneity.
• Use full-length 16S rRNA gene copies from whole-genome assemblies (in 'complete' stage) rather than partial sequences available from the public genetic databases to construct species-specific concatenated 16S rRNA libraries and further downstream analysis.
• Distinct four 16S rRNA gene copies cover all the Parsim-Info variable sites and can be used to construct a concatenated species-specific reference library.
• The total alignment score can be considered if the query sequence shows more or less the same percent identity with multiple species.
• Do not rely only on sequence similarity; make a final decision based on the phylogenetic inference.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)