Keywords
sample size estimation; longitudinal study; repeated measure analysis; univariate analysis
This article is included in the Manipal Academy of Higher Education gateway.
sample size estimation; longitudinal study; repeated measure analysis; univariate analysis
The introduction and background section has been updated with respect to adding the importance of precision and some spell correction. The ‘Notation and Framework’ section has been updated with the normal approximation assumption. The notation for null and alternative hypothesis as well as notation for sample estimates has been updated under the methods sections. More clarification has been added to the ‘Calculation of sample size section’ regarding timepoints, effect used and the derived sample size. Few appendices have been added to explain the theoretically phenomenon that was observed in the comparison of sample sizes with different correlation and different contrast types. We have also added references to a book and R packages for further exploration on power analysis in longitudinal studies.
See the authors' detailed response to the review by Kiranmoy Das
See the authors' detailed response to the review by Ronald Geskus
See the authors' detailed response to the review by Simon Vandekar
Understanding the concept of “sample size” is crucial for anyone involved in scientific research or clinical trials. The sample size refers to the number of subjects selected or observed in an experiment. This sample is a subset of the entire target population, which includes all individuals relevant to the study. For instance, in a study evaluating a new drug for type II diabetes, the target population would consist of all individuals suffering from this condition.
The sample size significantly influences the precision of our estimates and the study’s power, which is probability of correctly rejecting the null hypothesis when it is false. Essentially, higher power means a greater chance of detecting a true effect.
Two primary factors impact the power of a study: the sample size and the effect size. A larger sample size generally increases the study’s power, enhancing our ability to draw accurate conclusions. Effect size, on the other hand, measures the magnitude of the difference or relationship being studied between the groups.
In clinical trials, carefully calculating the sample size is essential. Correctly calculated sample size ensures that the study is adequately powered to meet its objectives, providing reliable and meaningful results. This meticulous planning is fundamental to advancing medical knowledge and improving patient care.
By paying close attention to sample size, researchers can design robust experiments that yield trustworthy insights, contributing to scientific progress and better health outcomes.
As an illustration, consider a study to compare the performance of a professional athlete taking a particular protein shake versus athletes who do not consume any special protein shakes. Narrowing down attention to a portion of the wider group is essential to enable tracking of the eating habits of every elite athlete in the world. Suppose this entails choosing one hundred professional athletes for our study at random; in this case, one hundred would be the sample size. Based on the data gathered from a sample of one hundred elite athletes, the study’s findings potentially characterize the population of all athletes in the sports industry. Lack of full coverage of the target population would result in the study’s outcome having a margin of error. Sampling error1 refers to the random variation between a sample estimate and the true population parameter due to observing only a subset of the population. It reduces the precision of an estimator (e.g., widening confidence intervals), which is crucial for generalizing results to the target population (e.g., all professional athletes). Greater precision enhances confidence in results, enabling more accurate analysis and informed decision-making.
Although sampling error cannot be completely eliminated, it can be reduced.2 A larger sample typically has a narrower margin of error. We require an appropriate sample size to examine and provide an accurate picture of the effects of protein shake consumption on performance. Note that increasing the sample size will help in reducing the sampling error but it does not address the non-sampling errors.
Longitudinal studies typically require longer follow-up periods but offer important advantages, including the assessment of temporal trends and stronger support for causal inference. While numerous longitudinal studies are available in literature, the purpose of this section is not to provide a comprehensive review of such research. Rather, it aims to illustrate that even in well-established and prestigious journals, longitudinal studies often provide limited detail regarding sample size calculation.
To examine how sample size calculations are reported in applied longitudinal studies, we conducted a targeted search of major bibliographic databases, including ‘Scopus’, ‘Web of Science’, ‘PubMed’, ‘ScienceDirect’, and ‘Google Scholar’ using a range of key terms: “designing clinical trials”, “sample size calculation”, “longitudinal studies”, “randomized trials” and “repeated measures”. This targeted review indicated that detailed descriptions of sample size calculation were frequently limited or absent in the reported methods of published longitudinal studies, as illustrated by selected examples.3,4 In contrast, methodological papers providing statistical formulae and frameworks for sample size determination in longitudinal designs are well documented in the literature.5,6 Additional examples from applied studies,7–11 discussed in the following paragraph, further highlight this recurring shortfall in reporting practice.1–3
Basagana, Liao and Spiegelman7 published a study in which the power as well as the sample size are discussed for time-varying exposures, but how this is practically applied to a longitudinal study design and its outcome is undocumented in published papers. Pourhoseingholi et al.,8 and Karimollah9 both published about the importance of various components for calculating the sample size in medical studies or clinical trials where often there would be more than one post baseline assessment, but sample size calculation is shown assuming single post baseline assessment. Manja and Lakshminrusimha published a two-part study10,11 which does give a good explanation on clinical research design, but sample size is not discussed in detail.
Most of the published studies which have assessments at multiple time points calculate the sample size based on the change from study end time point to baseline whereas a smaller number of papers emphasize on the use of multiple time points into consideration for calculating the sample size.5–7
The need for this research was prompted by this lack of proper usage of sample size calculation for longitudinal studies and to further explore which method for sample size calculation should be used in a longitudinal study resulting in correlated outcome data.
A wide range of methodologies and software tools for sample size and power calculation in longitudinal studies already exist12,13 (e.g., Diggle et al.; Ahn et al.; [R package references]). The present work does not seek to replace these approaches, but rather to complement them by explicitly contrasting single-time-point and multiple-time-point designs under simple and commonly assumed correlation structures.
To explore the variation in sample size by considering multiple time point assessment versus the change from baseline to a single endpoint.
Consider an experiment for testing certain hypothesis with parallel group design, having two independent groups to be treated under different scenarios to compare the outcome between the two groups. In our study we would consider the objective of comparison of two drugs.
Let ( ) be the outcome of interest at (j = 1, 2, 3, … , t) time point for the (i = 1, 2, 3, … , n) patient in the two groups.
For the parallel group design, these 2n patients will be divided into two groups with 1: 1 ratio where one arm is assigned to receive the test drug, and the other arm is assigned to receive the comparator drug.
Let & be the population mean for the test drug and comparator drug, respectively.
Let & be the sample mean outcome for the test drug and comparator drug respectively. The distribution of & can be approximated as and respectively under common standard deviation assumption. Under standard regularity conditions, the sampling distribution of the sample mean is approximately normal, with the accuracy of this approximation improving as the sample size increases, in accordance with the central limit theorem.14
Change at single post-baseline assessment (single time assessment analysis)
In a parallel group design study with two arms of equal size let the hypothesis be set as:
, no difference between the effects of test drug and comparator drug.
Vs
, test drug effect is greater than comparator and δ, δ > 0 is the expected difference.
The test statistic assuming known common standard deviation ‘ ’ (estimated from previous clinical trial data with same molecule which could be phase1, phase2 trials for same indication or pivotal trials with same molecule for different indication) for both arms will be given by
Now if is true ( ), then , else if is true (i.e., ) , then T will still follow Gaussian distribution but with a mean greater than zero.
If Type II error is denoted by then power will be simply and power is the probability to reject when is true. In probability equation it could be written as
Now in any study we would be looking for an inequality as below.
Upon solving Equation 5, we get15
Here, n is the sample size required per arm. We will use these formula in calculating the sample size for single post baseline time point analysis.
Post baseline assessment at multiple timepoints (multiple time points analysis)
In a parallel group design study with two arms of equal size and assessments taken at ‘t’ time points,
Let be the contrast to be tested for the hypothesis and let
With the hypothesis to be tested as:
, there is no difference between the effects of test and comparator drug.
, test drug is having larger effect as compared to comparator.
Where is the transpose of
& are the mean effect in arm one and arm two at time point ‘i’ respectively in a study with ‘t’ time points. can take any value depending on the hypothesis we want to evaluate.
For example, if we want to see the difference between two drugs when t = 2, then and the resulting will be
The variance-covariance matrix (estimated from previous clinical trial data with same molecule which could be phase1, phase2 trials for same indication or pivotal trials with same molecule for different indication), assumed to be known and similar between the two arms is given by
Where is the variance at time point ‘i’ and represents the covariance between time point ‘i’ and ‘j’.
The test statistic is given by
Where, and denote the sample-based estimators of the corresponding population quantities, such that the resulting expression constitutes a valid test statistic such that
with & being the sample mean at time ‘i’ in both the arms respectively.
Solving for using Equation 9, we get
Using Equation 10 in Equation 8 for T we get
Now, if we follow similar steps as we did in single time point analysis above, we get the following inequality.
And solving Equation 12, we get12
= common variance(assumed known)in the two groups at timepoint ‘i’
= common covariance(assumed known) in the two groups between timepoint ‘i’ and ‘j’.
= contrast applied at timepoint ‘i’ and ‘t’ represents the number of time points.
We will use the formulae specified in Equation 13 to calculate the sample size for multiple time point analysis.
Consider a 3-year study with assessments at baseline, 3 months, 6 months, 9 months, 12 months, 15 months, 18 months, 24 months, 30 months, 36 months. Appropriate sample size was calculated for multi-timepoint and single timepoint cases with different scenarios to achieve a difference of 0.9 points at 36 months between two treatment groups with an increasing trend from baseline. The different scenarios considered were with respect to equally spaced time points. Below table shows the timepoints selected for each scenario under longitudinal data sample size calculation. For single timepoint assessment only the baseline and 36 months were used.
A common standard deviation (SD) of 3.6 points was used; with type I error rate chosen as 5% and the power set at 85%. The effect size and standard deviation used here are observed in a real study.16 This was a three-year study with primary endpoint assessment at the end of year 3, but the sample size calculation in this study was done based on single time point. Since this study failed to recruit the expected number of patients and had a lot of missing data, the characteristics up until the second year’s evaluation were utilized because they featured a balanced number of patients in both treatment groups and had consistent measurements.
We considered a two-arm parallel group scenario with assessment at baseline and 36 months to assess the change from baseline in absolute scale. Using the formulae in Equation 6 above for single timepoint analysis to test the hypothesis, vs which is the expected difference), the sample size required per arm was 287 cases to achieve a difference of 0.9 points at the end of 36 months between two treatment groups with a common standard deviation (SD) of 3.6 points choosing type I error rate as 5% and the power set at 85% to show statistical significance.
Here again we considered two arm parallel groups with multiple timepoints and for studying we investigated six cases i.e., three, four, five, six, eight, and ten timepoints. Each of these cases correspond to multiple assessments including baseline. Three timepoints corresponded to the case with one baseline and two post baseline assessments, four timepoints corresponded to the case with one baseline and three post baseline assessments, five timepoints corresponded to the case with one baseline and four post baseline assessments, similarly other cases corresponded to one visit of baseline and remaining visits from pot baseline assessments.
Figure 1 and Figure 2 represents each of these cases as a line in the plot under different contrast types and correlation structures.
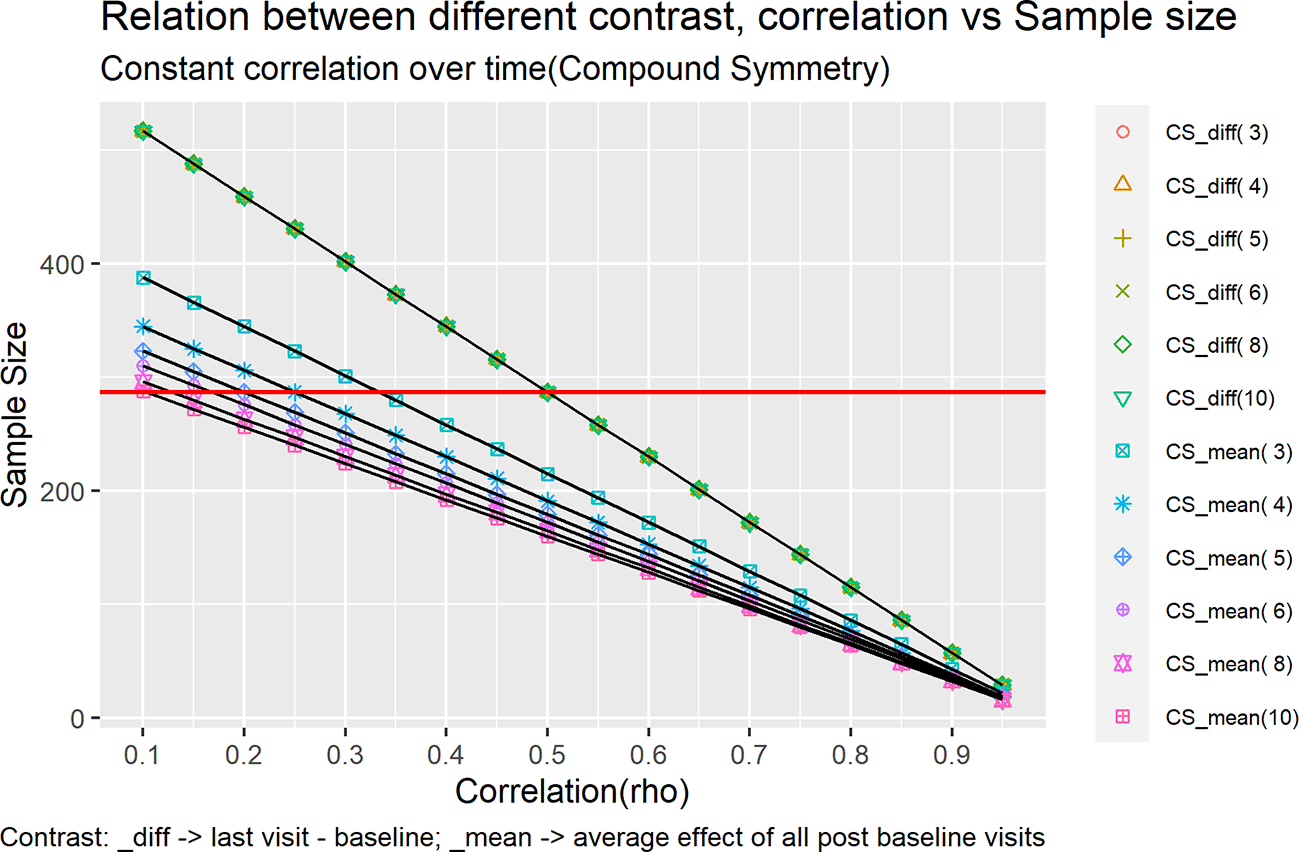
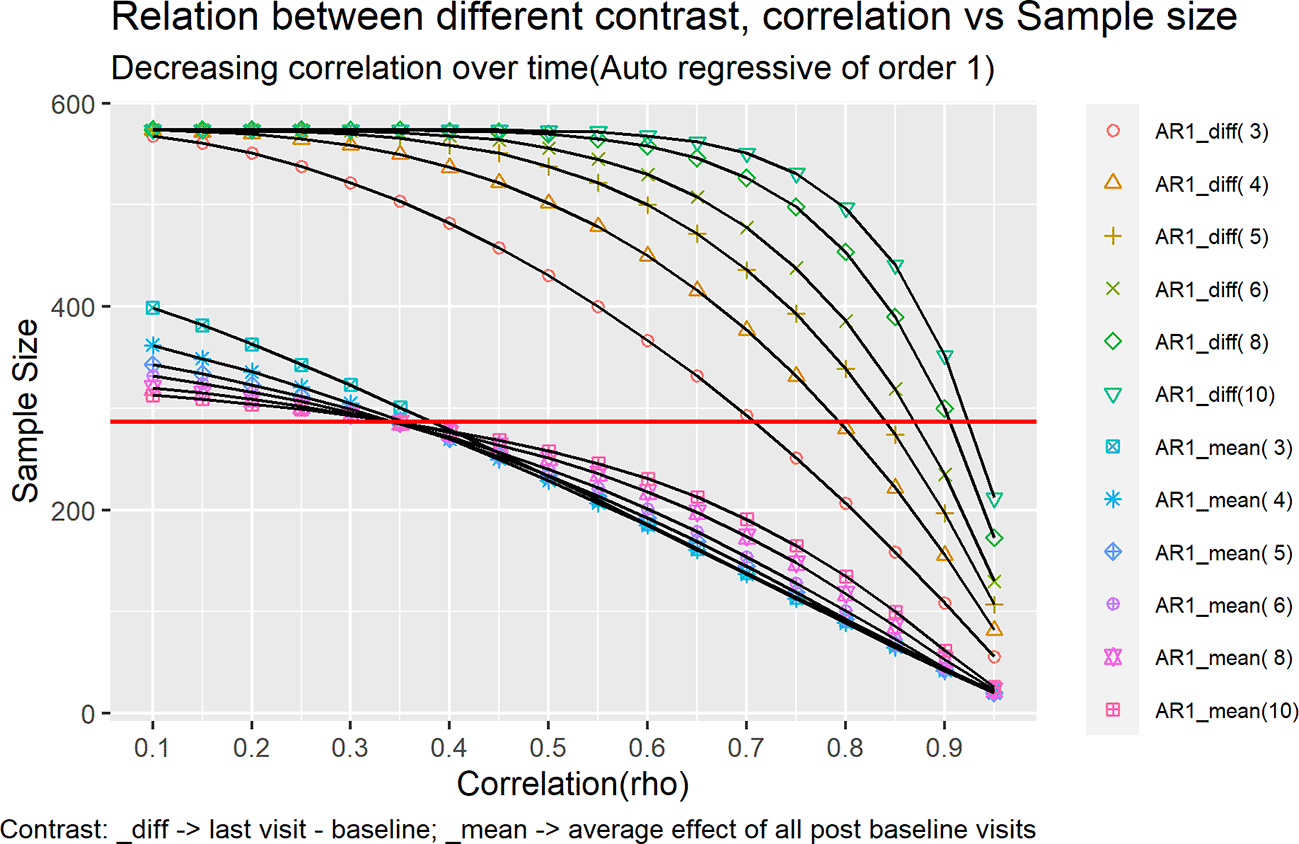
Keeping the SD as 3.6 we tried to vary over two different correlation structures:
Compound symmetry (CS)
Compound symmetry just means that all the variances are equal and all the covariances are equal. So, the same variance and covariance are used for all subjects. In compound symmetry the covariances across the subjects and the variances (pooled within the group) of the different repeated measures are homogeneous.
Where σ2 is the common variance assumed to be similar over time and ρ is the assumed correlation. The order of variance covariance matrix will be × , where ‘t’ is the number of time points. Generally, σ2 and ρ are estimated from previous clinical trial data with same molecule which could be phase1, phase2 trials for same indication or pivotal trials with same molecule for different indication.
Discrete Auto regressive of order 1(AR1)
This is the homogeneous variance first-order autoregressive structure. Any two elements that are adjacent have a correlation that is equal to rho (ρ), those separated by a third will have correlation ρ2, and so on. rho is restricted such that –1< ρ <1.
Where σ2 is the common variance assumed to be similar over time and ρ is the assumed correlation. The order of variance covariance matrix will be × , where ‘t’ is the number of time points. Generally, σ2 and ρ are estimated from previous clinical trial data with same molecule which could be phase1, phase2 trials for same indication or pivotal trials with same molecule for different indication.
Also, we considered different scenarios of how we want to analyze the results at the end as different contrasts as described below.
We investigated two types of contrasts.
1. Time-related contrasts i.e., mean over time (mean contrast).
Rationale: This contrast is more applicable in the situation where interest lies in observing the mean treatment effect on the subject’s disease condition over the period of time the subject is exposed to a prescribed dosing regimen compared to baseline.
This will be labelled in the legend of Figure 1 as CS_mean(i) and in the legend of Figure 2 as AR1_mean(i). For example, for five timepoints the contrast would look like [-1, ¼, ¼, ¼, ¼].
The mean-over-time contrast with equal weights was chosen to estimate the average treatment effect across follow-up, which is clinically meaningful in chronic disease settings and does not rely on the treatment effect following a strictly linear pattern over time.
2. Mean difference (change at last time point from baseline) (diff contrast).
Rationale: This contrast is more applicable in the situation where the interest lies in observing the treatment effect once the subject is exposed to a prescribed dosing regimen and what is the change at the end of the treatment exposure period in the subject’s disease condition. Here the total effect at the end of the treatment course as compared to the baseline is of interest.
This will be labelled in the legend of Figure 1 as CS_diff(i) and in the legend of Figure 2 as AR1_diff(i). For example, for five timepoints the contrast would look like [-1, 0, 0, 0, 1].
The sample size was calculated for correlation ranging from 0.1 – 0.95 with intervals of 0.05 for both the plots Figure 1 and Figure 2, using the formulas outlined in Equation 13.
All the trend lines for mean difference type of contrast overlaps each other. For mean difference type of contrast, the sample size does not change for an increase/decrease in the number of visits. It changes with the correlation i.e., highly correlated (rho > 0.5) timepoints would need less sample size as compared to low correlated timepoints. Also, for correlation = 0.5 the multiple assessment sample size coincides with that of single time point assessment.
However, the sample size does vary when the contrast is set to mean over time. Multiple time point assessments with more timepoints require less sample size as compared to that of multiple time point assessment with less time points for example, the multiple time point assessment with three timepoint requires 86 per arm with correlation 0.8 and the multiple time point assessment with ten timepoints requires 64 per arm. On the same lines the multiple time point assessment with three timepoints requires 258 per arm with correlation 0.4 and the multiple time point assessment with ten timepoints requires 192 per arm. This trend shows that under compound symmetry, the efficiency gain from repeated measurement depends on the within subject correlation. The required sample size decreases as the correlation between timepoints increases. However, under CS, at moderate correlation levels (ρ = 0.5), this gain may be neutral, whereas lower correlations reduce efficiency and increase the required sample size.
Under mean over time contrast the multiple time point assessment requires lower sample size as compared to single time point assessment (287 per arm) for correlation greater than 0.35 and the sample size increases as correlation goes below 0.35. For correlation 0.35 the sample size coincides with that of single time point assessment for all the cases except the case of three timepoints which requires slightly higher sample number.
Whereas for mean difference contrast the multiple time point assessment requires lower sample size as compared to single time point assessment (287 per arm) for correlation greater than 0.7 but requires higher sample size for correlation less than 0.7.
The trend changes shape for mean difference contrast vs mean over time contrast. Also, at certain point the increase in sample size attenuates for example, in case of mean difference type contrast with ten time points the sample size required does not change when correlation drops below 0.55.
One of the hurdles in considering the longitudinal methodology for sample size calculation is the assumption on the covariance matrix. It is often easy to estimate the variance of single timepoint as compared to estimating the variance-covariance matrix for multiple time points.
The above derivations were done for trial design with parallel group, 1:1 ratio and two arms. If the ratio changes or if we have more than two arms or if the design is crossover, then the effective overall sample size would get effected in both the cases i.e., sample size with single time point as well as sample size with multiple timepoints, but the trend would remain the same as shown above in the figures and the results will still hold good. Similar trends should hold for other variance – covariance structures though they have not been simulated here.
Sample size changes depending on the analysis type and the data collected. Both the graphs in Figure 1 and Figure 2 in this study reveal that if response is assessed at multiple timepoints and the correlation between the paired observations is high (> 0.6) then one should consider using repeated measures analysis and consequently determine the size of the sample that is based on the multiple time points scenario which results in lower sample size requirement as compared to the sample size derived assuming single timepoint response assessment. This would reduce the cost, resources, and time in conducting the experiment fastening the new drug development. Also, repeated measures analyses will not drop the patients in which they have certain missing data as compared to single point analysis where the patient will be dropped if the response is missing and hence may help in retaining the power.
Under a compound symmetry (CS) covariance structure, the required sample size for the mean over time contrast decreases when information from a greater number of time points is available, reflecting the additional information gained from repeated measurements. In contrast, for the mean diff contrast, which is based on a comparison between two time points only, the required sample size remains unchanged regardless of the total number of additional time points observed.
Under an AR(1) covariance structure, different patterns are observed. For the mean over time contrast, the required sample size increases as more time points are included when the within-subject correlation exceeds ρ = 0.35, whereas for ρ ≤ 0.35 the required sample size decreases with the inclusion of additional time points. For the mean diff contrast, the required sample size with information on fewer timepoints is lower as compared to the sample size required when we have information available at more timepoints (Figure 2).
These patterns are not universal but depend on the assumed correlation structure and the magnitude of the mean difference at the chosen time point. Further investigation is warranted to better understand how these relationships vary across alternative covariance structures and effect trajectories.
Sample size derivation using longitudinal design method for studies with multiple assessments can be considered of substantial benefit in cost and time although the challenge of estimating the variance-covariance matrix remains. When within subject correlations are low, averaging treatment effects across multiple time points may increase estimator variance, thereby reducing efficiency relative to a single and well-chosen time point comparison. Also, to be noted that the distance between the timepoints is not taken into consideration by the sample size with multiple timepoints derivation method which could be a topic of further research.
The sample size derivation for multiple timepoint assessment Equation 13 can directly be used by the readers using their treatment effect, variance covariance matrix and the contrast of their choice. To ease this, we have also provided the link to R code under software availability section where inputs for treatment effect, variance covariance matrix and contrast can be given to the function to obtain the sample size required. To explore more on power analysis in longitudinal studies one may refer to the book “Sample Size Calculations for Clustered and Longitudinal Outcomes in Clinical Research”.13 One could also make use of the R packages for different cases of longitudinal power analyses provided under the ‘The R Journal’ article “Power and Sample Size for Longitudinal Models in R”.17
Software available from: The Comprehensive R Archive Network (https://cran.r-project.org/)
Source code available from: https://github.com/Sarfaraz-Sayyed/Sample-Size-Variation.
Archived source code at time of publication: https://zenodo.org/badge/latestdoi/547747570.18
Archived source code at time of revision: Sample Size Variation (zenodo.org)
License: MIT License.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
 ), as
), as  ” and “
” and “ , where
, where  , and δ represents the expected difference. This definition clarifies the role of δ in the sample size derivation and avoids ambiguous statements. The corresponding revision has been made in the Methods section and aligns with Equation (2) accordingly.
, and δ represents the expected difference. This definition clarifies the role of δ in the sample size derivation and avoids ambiguous statements. The corresponding revision has been made in the Methods section and aligns with Equation (2) accordingly. ) increases rather than decreasing.
) increases rather than decreasing.
Comments on this article Comments (0)