Keywords
SARS-CoV-2, Diagnosis RT-qPCR, Adjuvant formulation, Primer and probe design, High performance computing
This article is included in the Pathogens gateway.
This article is included in the Emerging Diseases and Outbreaks gateway.
This article is included in the Coronavirus (COVID-19) collection.
SARS-CoV-2, Diagnosis RT-qPCR, Adjuvant formulation, Primer and probe design, High performance computing
This version has been substantially revised in response to the reviewer’s comments to improve clarity, methodological transparency, and consistency between the reported results and their interpretation. The manuscript now more explicitly presents the study as an exploratory and methodological contribution rather than as a clinical diagnostic validation study. The Abstract, Introduction, Results, Discussion, and Conclusions have been revised to moderate statements that could be interpreted as overstating the findings. In particular, claims regarding assay performance have been reformulated to more accurately reflect the scope of the dataset and to avoid implying broad clinical diagnostic superiority. The Methods section has been clarified, particularly with respect to the Ct-based performance analysis. This analysis is now described as an exploratory internal comparison based on predefined operational Ct criteria, and its limitations are stated more explicitly. The corresponding Results section has also been revised so that these metrics are presented as comparative descriptors of assay behaviour under the evaluated experimental conditions rather than as measures of absolute diagnostic accuracy. In addition, a dedicated Limitations section has been included to acknowledge the restricted sample size, the constraints associated with sample availability during the COVID-19 pandemic, the absence of independent external clinical validation, and the ethical and legal restrictions that limited access to additional clinical information. Finally, the Data Availability section has been expanded to improve transparency and reproducibility by more clearly describing the genomic datasets, sequence alignments, and analysis scripts deposited in public repositories.
See the authors' detailed response to the review by Andrew D. Beggs
See the authors' detailed response to the review by Juan Sebastian Quintero Barbosa
On January 23, 2020, the SARS-CoV-2 virus (Coronaviridae: Betacoronavirus: Severe acute respiratory syndrome-related coronavirus) was declared a public health emergency by the World Health Organization (WHO) International Health Regulations (IHR) Emergency Committee. At the time, the global public health authorities established that the spread of SARS-CoV-2 could be prevented if every nation adopted solid strategies for rapid and accurate disease detection (WHO, 2020).
Diagnostic methods based on gene-specific primers and probes for the detection of viruses via gene amplification include quantitative real-time polymerase chain reaction (RT-qPCR) or reverse transcription loop-mediated isothermal amplification (RT-LAMP), both of which can be conducted using oropharyngeal and nasopharyngeal swab samples from patients (Kevadiya et al., 2021). Among these, the former method is considered the most sensitive and accurate for the detection of SARS-CoV-2 and other viruses (Martín et al., 2021). This procedure could be effectively implemented due to the characterization of the viral genome, which encompasses approximately 10 genes (Zhu et al., 2020). Therefore, WHO authorized the Berlin protocol which relies on the following genes: ORF1ab, which encodes proteins that enable viral replication (Nur et al., 2015); Spike (S), which interacts with the receptor of the host’s angiotensin-converting enzyme 2 (ACE2) (Wan et al., 2020); and E, which encodes the structural envelop protein (Tahan et al., 2021). Other protocols such as the 2019-nCoV TaqMan RT-qPCR Kit authorized by the United States Centers for Disease Control and Prevention (CDC) utilize two regions of the N gene (N1-N2), which encode the nucleocapsid phosphoprotein (Navarathna et al., 2021). Moreover, both protocols use the RNase P gene as a control to assess the efficiency of the RT-qPCR protocol (WHO, 2009).
Nevertheless, RT-qPCR-based diagnosis is conducted on a per-gene basis and therefore its widespread implementation would be both impractical and costly. Therefore, multiplex RT-qPCR (e.g., the CDC kit or combined quantification of the ORF1ab and S genes) could be implemented as a promising approach to meet the current demand for accurate and cost-efficient diagnosis (Kudo et al., 2020). Sample pooling is another approach that could increase population-wide SARS-CoV-2 diagnosis rates and has therefore been approved and implemented in several countries (Grobe et al., 2021). However, sample numbers and sampling structure may limit the implementation of this strategy (Grobe et al., 2021).
Although RT-qPCR-based SARS-CoV-2 diagnostic tools are generally considered the gold standard for disease detection, several studies have determined that this approach is prone to return false-negative results due to gene mutations (which is often the case for the E and N genes) (Hasan et al., 2021; Tahan et al., 2021) or primer dimer formation (Jaeger et al., 2021). Another factor that affects RT-qPCR efficiency is the secondary structure of the RNA to be characterized/quantified (Hammerling et al., 2020).
Previous studies have demonstrated the role of RNA secondary structure in viral evolution and transcript regulation (Andrews et al., 2021; Huston et al., 2021; Rangan et al., 2020; Wacker et al., 2020). However, their potential impact on RT-qPCR performance in clinical samples remains insufficiently characterized. Furthermore, the effects of adjuvants such as dimethyl sulfoxide (DMSO) or ammonium salts on RT-qPCR efficiency are not fully understood (Kovarova & Draber, 2000).
In this context, we used High-Performance Computing (HPC) to compile a global dataset of SARS-CoV-2 genomes available up to December 2020 and selected a region of the ORF8 gene for evaluation in both single and multiplex RT-qPCR assays, alongside targets from the E and N genes. Additionally, we assessed the potential impact of adjusting denaturing solution composition and nucleotide proportions on RT-qPCR performance.
This study was designed as an exploratory and methodological approach to evaluate how RNA secondary structure and nucleotide composition may influence RT-qPCR assay performance under defined experimental conditions. The implications of these findings are discussed in the following sections.
This study was approved by the Research Ethics Committees of the Industrial University of Santander, the Chicamocha Clinic, and the Chicamocha Clinical Laboratory (L3C) (Bucaramanga, Santander, Colombia). The study participants were hospitalized patients diagnosed with SARS-CoV-2, individuals presenting to the emergency room, or volunteers. All participants provided written informed consent and voluntarily participated in our study. Further, all participants were kept anonymous and were informed that the present study was conducted strictly for research purposes and that its outcomes were not intended to serve as treatment or diagnosis.
Nasopharyngeal swab samples were acquired by L3C personnel. Two samples were obtained per study participant, one for clinical diagnosis, as authorized by the Ministry of Health and Social Protection of Colombia, and the other for our study. The samples were placed in Universal Transport Medium (UTM) and stored at −80°C until required for downstream analyses (Rogers et al., 2020). Samples were collected over a one-week period and packaged in accordance with applicable Colombian health regulations (Ministry of Health and Social Protection, 2020) prior to transport and processing. All samples were transferred to the Central Research Laboratory of the Faculty of Health Sciences at the Industrial University of Santander (LCI-FS-UIS) and maintained at 4°C during handling and processing. In all cases, written informed consent was obtained prior to sample collection, and documentation was properly verified and archived in accordance with institutional and national ethical guidelines.
Because sample collection was not conducted under a predefined experimental sampling framework but was instead constrained by voluntary participation and ethical requirements during the COVID-19 pandemic, an anonymized coding system was implemented to ensure participant confidentiality, in accordance with Resolution 8430 of 1993 and the applicable Colombian ethical framework (Ministry of Health and Social Protection, 1993). The alphanumeric code consisted of two digits corresponding to the participant, followed by “CO” (Colombia) and a consecutive number assigned to each sample.
Subsequently, samples were aliquoted in a Class II, Type A2 biosafety cabinet (Thermo Fisher Scientific). A total of three aliquots were obtained per sample, including one 200 μL aliquot used immediately for total RNA extraction and two 650 μL backup aliquots stored at −80°C.
To account for reported mutations in SARS-CoV-2 genomes that could affect primer and probe binding and potentially generate false-negative results, a region encoding the accessory protein ORF8 was selected for analysis. This region was chosen based on its predicted mutation profile, and monthly consensus sequences were generated to assess its identity pattern across newly available genomes. Publicly available SARS-CoV-2 genome sequences were obtained from the GenBank and GISAID databases (RRID:SCR_002760; RRID:SCR_018251) (Sayers et al., 2021; Shu & McCauley, 2017). To gather data from the GenBank database, a Python script (RRID:SCR_008394) was executed in the GUANE-1 High Performance and Scientific Computing Center of the Industrial University of Santander (SC3UIS) through the Entrez Programming Utilities interface using the Biopython package (RRID:SCR_013249; RRID:SCR_007173) (Cock et al., 2009; Geer et al., 2010), whereas all GISAID data were manually downloaded. Genomes with uncharacterized regions/nucleotides were discarded using another Python script (RRID:SCR_008394), which was used to conduct monthly FASTA sequence alignments using the MAFFT software (RRID:SCR_011811) (Katoh & Standley, 2013) coupled with previously described DNA loss model parameters (Martínez-Pérez et al., 2002, 2007). The consensus sequences were generated using BioEdit version 7.2 (RRID:SCR_007361) with a 100% threshold frequency (Hall, 1999).
Using the SARS-CoV-2 (NC_045512) reference genome and consensus sequences from January to April 2020 (Zhu et al., 2020), an approximately 150-base pair (bp) region of the ORF8 gene was selected based on the secondary structure of its RNA transcript, which in turn was predicted using the algorithms proposed by Zuker (Zuker & Jacobson, 1998) via the Mfold software (RRID:SCR_001360) (Zuker, 2003). Thus, the last set containing the codons of the central region of the ORF8 gene, which encodes the secretion protein ORF8, was chosen because it allows proper viral adhesion to the host cell (Chan et al., 2020). The obtained sequence was used as a template to create primers, a TaqMan FAM-BBQ probe, and substrate oligos for RT-qPCR. The specificity of the aforementioned molecules was confirmed via GenBank BLAST analyses (RRID:SCR_004870) (Ye et al., 2006). Genes E and N from the Berlin and CDC protocols were used as controls (Biotek, 2020; Corman et al., 2020).
All molecules were synthesized by Bioneer (Korea). The primers were purified via separation on a reverse-phase cartridge, whereas the probe and substrate oligos were purified via high-performance liquid chromatography (HPLC) and polyacrylamide gel electrophoresis (PAGE), respectively. The ORF8 RT-qPCR conditions were implemented as described by the Berlin protocol (Corman et al., 2020) using the aforementioned molecules coupled with the 2019-nCoV TaqMan RT-qPCR Kit (Norgen Biotek Corp) developed by the CDC (Biotek, 2020).
A:T and G:C ratios were calculated based on the monthly SARS-CoV-2 consensus sequences to obtain an average for each nucleotide. These averages were then used to determine the minimum concentrations of TEA (ABCAM-USA), DMSO (Scharlab-Spain), and dNTPs (100 mM each, Promega-USA) in molecular-grade ultrapure water (Promega-USA), in addition to the MgSO4 concentration recommended by the Berlin protocol.
Total RNA extraction was conducted using the MagMAX™ Viral/Pathogen II (MVP II) Nucleic Acid Isolation Kit (2000 RXNs) (Applied Biosystems-USA) using a KingFisher Duo Prime (5400110) DNA/RNA extraction system according to the manufacturer’s instructions (Thermo Fisher Scientific-USA) (Fang et al., 2007).
RT-qPCR was conducted using the ORF8-specific primers and probe designed herein, in addition to the E (Berlin protocol) and N genes (N1-N2; CDC protocol). The RNase P gene was used as an external control, as proposed by both of the aforementioned protocols. The reactions were conducted using the SuperScript III One-Step RT-qPCR System with Platinum Taq DNA Polymerase (Ref. 11732088; Invitrogen-USA) and the 2019-nCoV TaqMan RT-qPCR Kit (Ref. TM67100; Norgen-Biotek-Canada). Each reaction for each diagnostic system was conducted in either 15- or 25-μL reaction volumes consisting of 2 μL of patient-derived purified RNA, 2× One-Step RT-qPCR Master Mix, 2× nuclease-free buffer, and the respective primers/probe at the concentrations recommended by the CDC. Moreover, a denaturing stock solution was added to obtain a final concentration of 0.7% TEA, 0.2% DMSO, and 0.8 mMol MgSO4. The dNTP reagent had a final concentration of 12 mMol dATP-dTTP, 10 mMol dCTP-dGTP, and 0.8 mMol MgSO4. The reaction conditions were the following: 55°C for 15 minutes, 95°C for 3 minutes followed by 45 cycles of 95°C for 15 s and 58°C for 30 s for the SuperScript™ III One-Step RT-qPCR kit; and 95°C for 3 s followed by 55°C for 20 s for the 2019-nCoV TaqMan RT-qPCR kit. Fluorescence signals were quantified using a QuantStudio 1 Real-Time PCR System (No. A40427) in a 96-well 0.2 μL block (Thermo Fisher Scientific-USA).
The above-described procedure was conducted using two different Multiplex One-Step RT-qPCR protocols. In the first instance, the primers and probes for the E, ORF8, and N (N1) genes were mixed, whereas the other reaction was performed by mixing the N1 and N2 sets of the N gene. The RNase P gene was independently assessed in both cases. All reactions were performed as described above.
Single and multiplex RT-qPCR assays were compared based on Ct values obtained under identical experimental conditions. Comparative analyses were performed at both the individual primer/probe level and at the sample level, allowing evaluation of performance differences between assay formats.
To estimate sensitivity, specificity, Positive Predictive Value (PPV), and Negative Predictive Value (NPV) for the single and multiplex RT-qPCR assays, a custom script was implemented in R using the Caret package (https://github.com/GenomicUIS/Sensitivity-specificity-PPV-and-NPV-for-SARS-CoV-2.git) (Kuhn, 2008; R Core Team, 2020). All scripts and processed datasets used in these analyses are publicly available in the referenced repository.
Because this study was designed as an exploratory methodological study and not as a clinical diagnostic validation, performance estimates were derived from predefined Ct-based operational criteria applied uniformly across assays. For individual reactions, Ct values between 18 and 35 were considered within the operational positive range. Ct values outside this interval, including values >35 or <18, were conservatively classified as non-positive or indeterminate for this exploratory internal analysis.
Ct values were subsequently pooled and evaluated at the sample level. A sample was considered positive when at least one primer/probe set met the predefined operational Ct criterion. For multiplex RT-qPCR assays, the same Ct-based criteria were applied, but results were evaluated collectively at the sample level.
These metrics were used to compare assay behavior under the evaluated experimental conditions. Because no independent external clinical validation was available, and because access to additional clinical information was restricted by ethical and legal considerations, these estimates should not be interpreted as measures of absolute diagnostic accuracy.
A total of 19,317 genomes were retrieved from the GenBank database from January to October 2020 based on our search criteria, whereas 107,259 complete genomes were obtained from the GISAID database between January and December 2020. In both datasets, the number of available sequences increased progressively over time. However, the frequency of base substitutions in the monthly consensus sequences for the E, ORF8, and N genes was higher in the GISAID dataset compared to GenBank ( Figure 1).
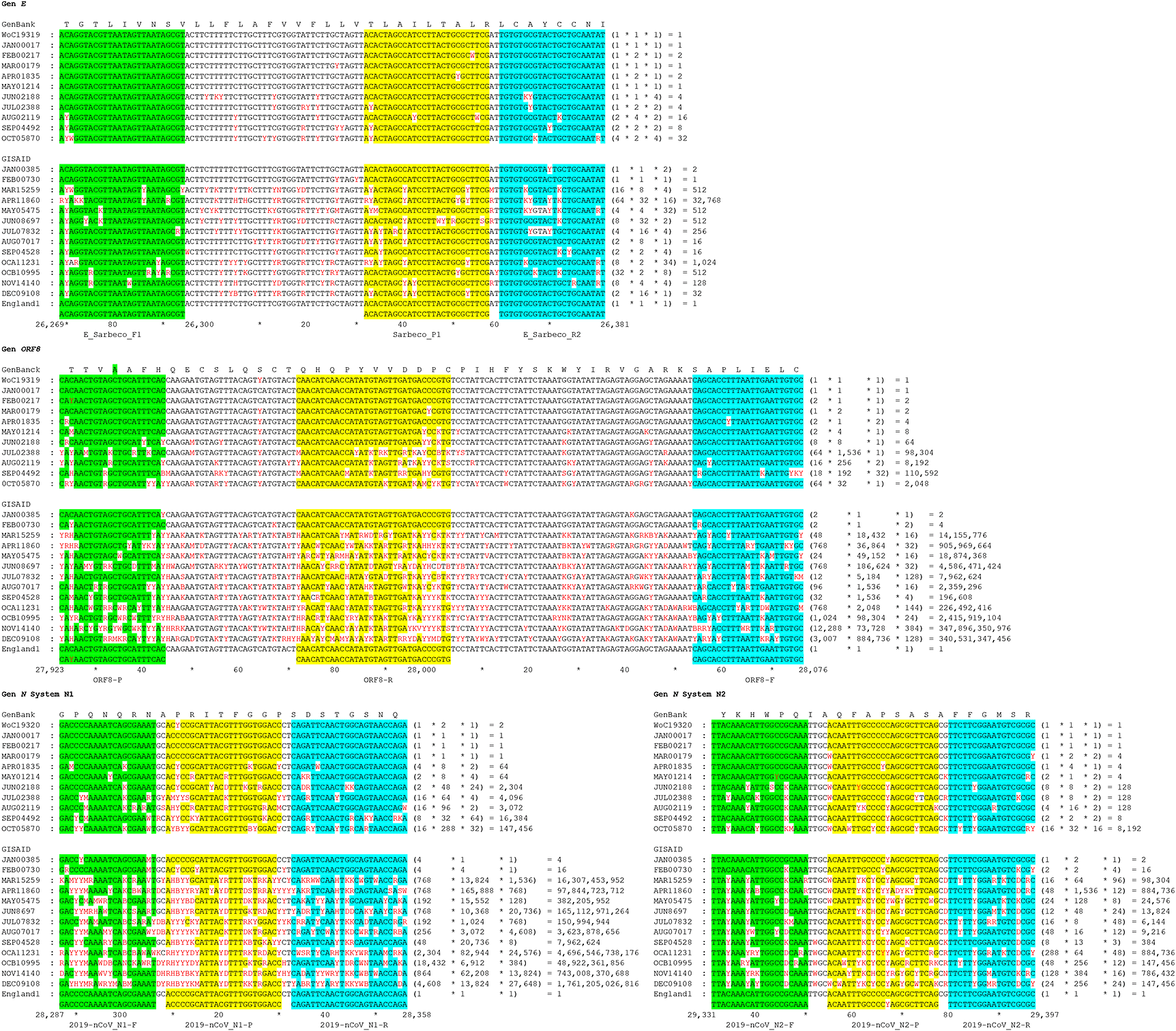
Nucleotide sequences are indicated as follows: forward primers are shown in green boxes, reverse primers in blue boxes, and probes in yellow boxes. Conceptual translation is displayed above each alignment. Numbers indicate nucleotide position within the sequence, and nucleotide combinations at each position follow international nomenclature. The nucleotide positions relative to the SARS-CoV-2 reference genome are shown in the lower consensus alignment.
The region of the E gene employed herein exhibited a 113 bp length, whereas the amplicons of the N1-N2 system of the N gene were 72 and 67 bp in length, respectively. All regions exhibited loop-bubble structures. A 154 bp region within the first half of the ORF8 gene also presented a loop-bubble structure similar to those observed in the E and N genes. The third loop of the N1 system and the second loop of the ORF8 gene were formed by four and seven canonical base pairings, respectively. These structures are comparable to scorpion-type primer or probe configurations used in RT-qPCR; however, such loop-bubble structures are typically formed by 2–6 canonical base pairings ( Figure 2). Table 1 summarizes the primer sequences used in this study.
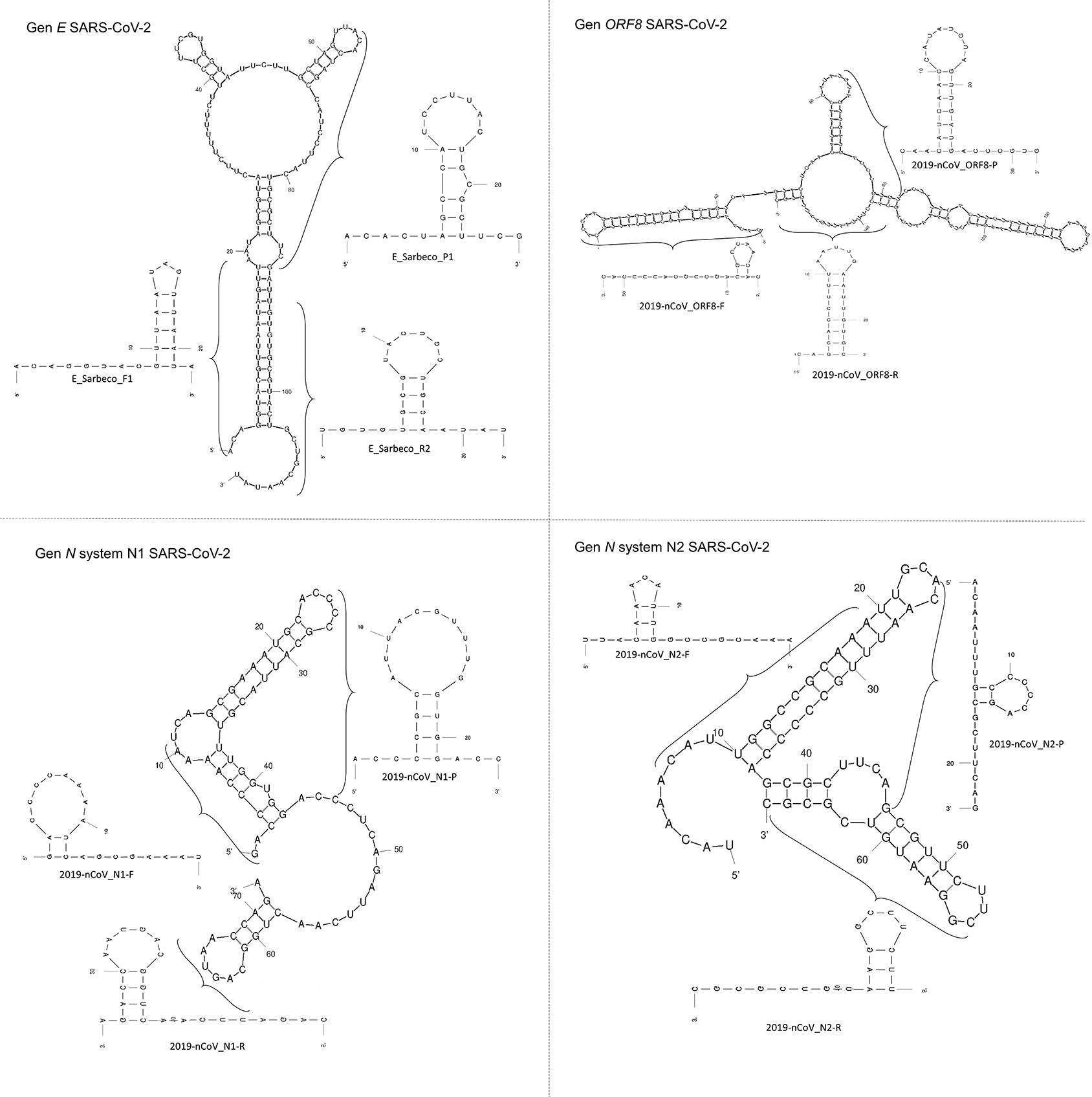
Stem and stem–loop structures are observed in both the 5′ and 3′ orientations. Numbers indicate the number of nucleotides in each structure, and parentheses delineate individual primers and probes within each segment.
| Gen | Type | Code | Sequence | Total nmole | Reference |
|---|---|---|---|---|---|
| ORF8 | Primer Forward | FwO820CoV | CAYAACTGTAGCTGCATTTCAC | 24.3 | This work |
| Primer Reverse | RvO820CoV | GCACAATTCAATTAAAGGTGCTG | 21.9 | ||
| Probe | TqMO820CoV | FAM-CAACATCAACCATATGTAGTTGATGACCCGTG-BBQ | 8.7 | ||
| Substrate Oligonucleotide | TrGtO8 | CAYAACTGTAGCTGCATTTCACCAAGAATGTAGTTTACAG TCATGTACTCAACATCAACCATATGTAGTTGATGACCCGT GTCCTATTCACTTCTATTCTAAATGGTATATTAGAGTAGG AGCTAGAAAATCAGCACCTTTAATTGAATTGTGC | 0.3 | ||
| E | Primer Forward | E_Sarbeco_F1 | ACAGGTACGTTAATAGTTAATAGCGT | 34.0 | (Corman et al., 2020) |
| Primer Reverse | E_Sarbeco_R1 | ATATTGCAGCAGTACGCACACA | 49.8 | ||
| Probe | E_Sarbeco_P1 | FAM-ACACTAGCCATCCTTACTGCGCTTCG-BBQ | 22.5 | ||
| N | Primer Forward | 2019-nCoV_N1-F | GACCCCAAAATCAGCGAAAT | 22.5 | (Biotek, 2020) |
| Primer Reverse | 2019-nCoV_N1-R | TCTGGTTACTGCCAGTTGAATCTG | 22.5 | ||
| Probe | 2019-nCoV_N1-P | FAM-ACCCCGCATTACGTTTGGTGGACC-BHQ | 22.5 | ||
| Primer Forward | 2019-nCoV_N2-F | TTACAAACATTGGCCGCAAA | 22.5 | ||
| Primer Reverse | 2019-nCoV_N2-R | GCGCGACATTCCGAAGAA | 22.5 | ||
| Probe | 2019-nCoV_N2-P | FAM-ACAATTTGCCCCCAGCGCTTCAG-BHQ | 22.5 | ||
| RNase P | Primer Forward | RP-F | AGATTTGGACCTGCGAGCG | 54.3 | (Corman et al., 2020) |
| Primer Reverse | RP-R | GAGCGGCTGTCTCCACAAGT | 53.2 | ||
| Probe | RP-P | FAM–TTCTGACCTGAAGGCTCTGCGCG–BHQ | 12.8 |
A total of 49 samples were collected between October 25, 2020, and January 21, 2021, of which 22 tested positive and 27 tested negative. Samples collected in October were used to evaluate the detection of SARS-CoV-2 using the E, ORF8, and N gene targets by RT-qPCR ( Table 2, Figure 3). Based on the secondary structure and nucleotide composition analyses, which indicated an A:T ratio between 63% and 70% across consensus sequences, denaturing and dNTP solutions were incorporated into the reactions (see Methods for details). Under these conditions, changes in Ct values were observed compared to the commercial procedures, although variability was detected among samples ( Table 2, Figure 3).
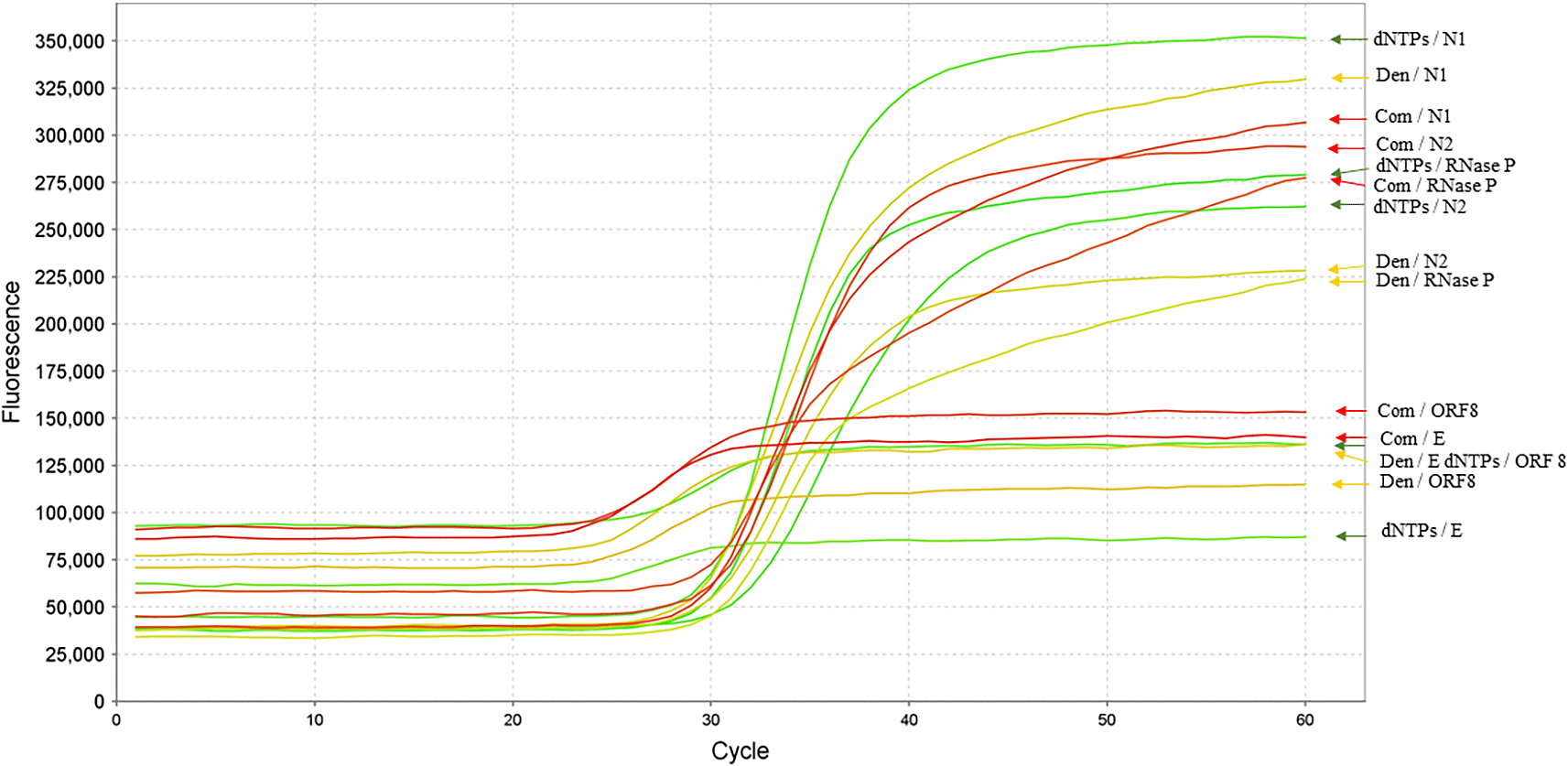
RNA extracted from patient samples was analyzed using primers and probes targeting the E gene (WHO protocol), ORF8 (this study), and the N gene (N1–N2; CDC protocol). Green, yellow, and red arrows indicate amplification curves obtained using denaturing solution (Den), dNTP-adjusted conditions, and commercial kits, respectively. The RNase P gene was used as an internal control.
A decrease in amplification performance was observed in samples collected from December onward. This effect was first detected in the E gene system, followed by the N2 system and subsequently ORF8. This pattern was evaluated using ten samples collected between December 14 and 21, 2020. The observed results were classified into three outcomes depending on the detection system: (1) positive results were obtained following the manufacturer’s protocol with the addition of the denaturing solution; (2) positive results were obtained only in the presence of the denaturing solution; and (3) positive results were obtained only in the absence of the denaturing solution ( Table 2).
Analysis of global SARS-CoV-2 mutation patterns using GenBank and GISAID databases showed that the number of mutations increased from March 2020 onward relative to the reference genome (GenBank accession: NC_045512.2). This trend was particularly evident in the quencher and forward primer regions of the ORF8 and N genes. The number of potential primer and probe combinations also increased over time, with a marked rise observed from November onward in the GISAID dataset. Higher values were observed in December. For example, the ORF8 system and the N1 set exhibited 3.4×1011 and 1.7×1012 possible combinations, respectively, whereas the N2 set and the E gene exhibited 1.4×105 and 32 combinations ( Figure 1).
Multiplex RT-qPCR controls using the E, ORF8, and N (N1) systems with positive samples 02 and 03 produced amplification profiles consistent with detection. Similar patterns were observed in samples 05 and 06 collected in October 2020. Under the same reaction conditions, the addition of the denaturing solution was associated with decreased Ct values in samples collected in October, whereas increased Ct values were observed in samples collected in November. In contrast, the dNTP solution exhibited an opposite trend ( Table 3, Figure 4).
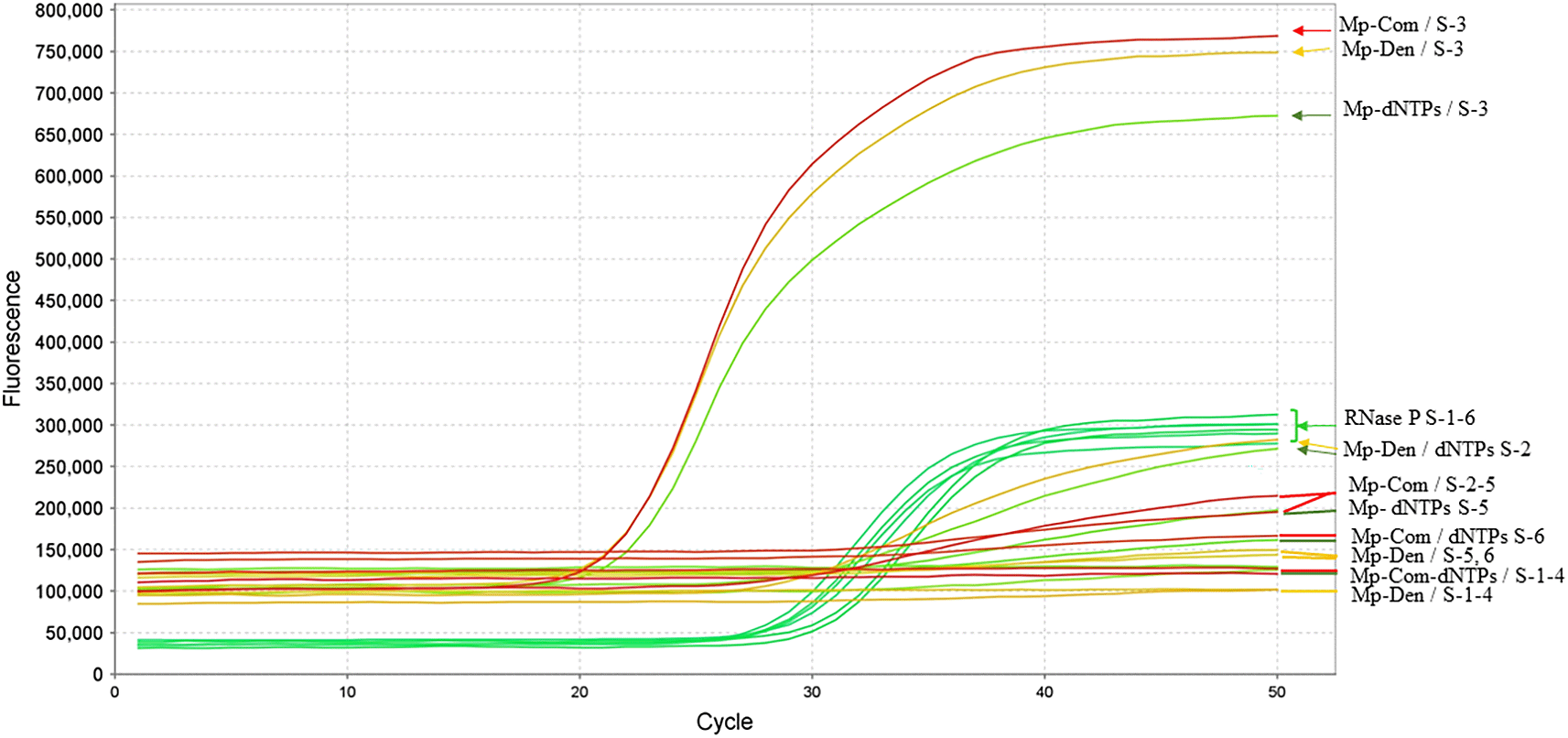
RNA samples from six patients are indicated by their corresponding identifiers. For each curve, the first number in the nomenclature corresponds to the upper panel, and the second to the lower panel. The RNase P gene was used as an internal control. Arrows and nomenclature are consistent with those described in Figure 3.
The influence of the denaturing and dNTP solutions was further evaluated using nine samples collected between December 4 and 7, 2020, and January 14 and 15, 2021, using the N1–N2 primers and probe described by the CDC. Six samples exhibited the same pattern described above, whereas three samples tested negative based on both amplification curves and Ct values ( Table 3, Figure 5). The multiplex RT-qPCR assay detected SARS-CoV-2 in samples collected between January 19 and 29, 2021 ( Table 3).
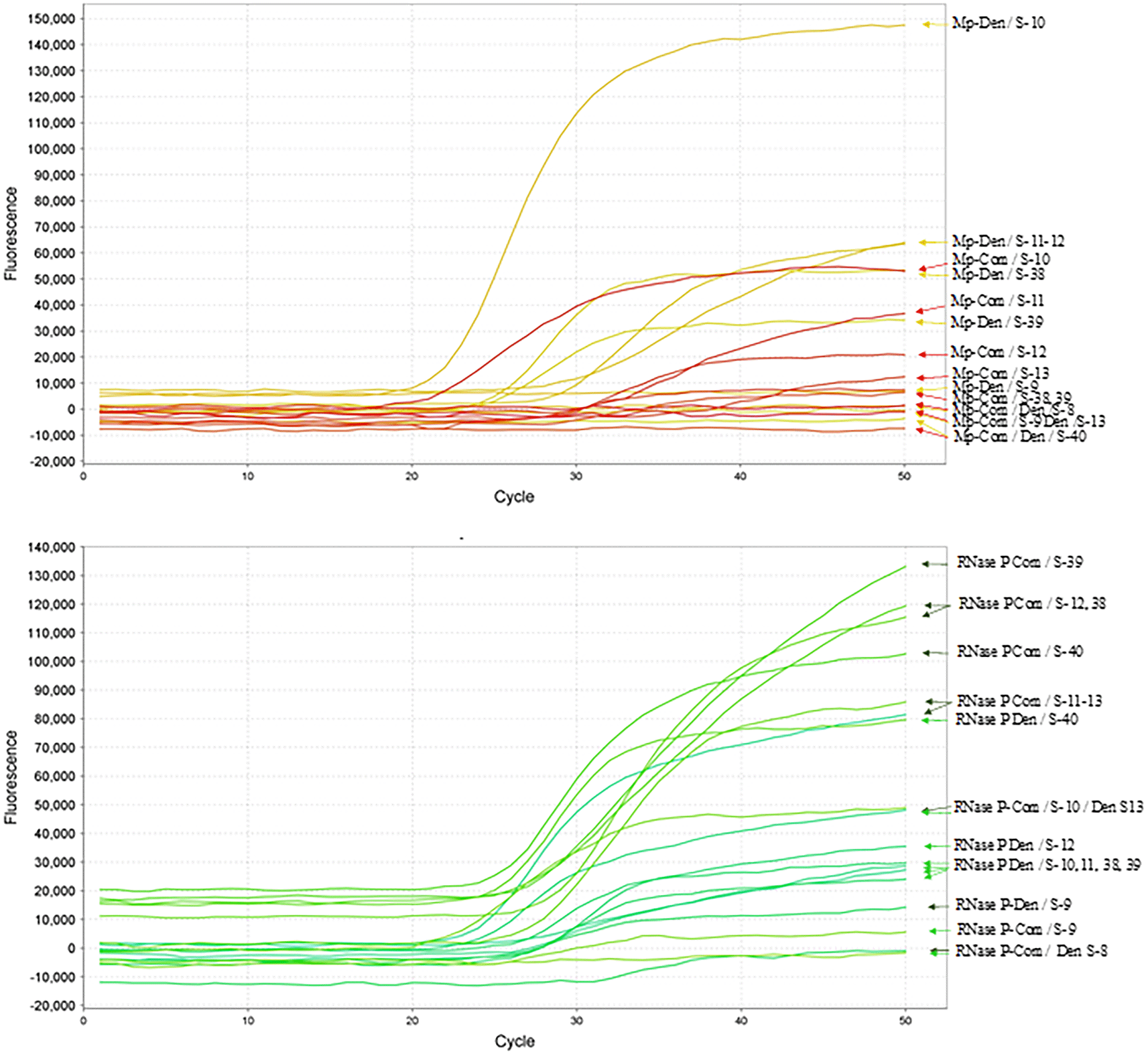
CDC-described primers and probes were used in combination with the denaturing reagent. The RNA samples from 9 patients are indicated with the respective numbers. The multiplex curves corresponding to N1-N2 are indicated at the top, whereas the results corresponding to the RNase P primers and probe used as controls are indicated below. The abbreviations and nomenclature are the same as in Figure 3.
Under the predefined Ct-based classification framework, the denaturing solution was associated with higher apparent sensitivity values than the commercial solution in single RT-qPCR assays, whereas the commercial solution showed higher apparent specificity values. In multiplex RT-qPCR assays, no major differences in apparent sensitivity were observed between commercial and denaturing conditions.
In multiplex reactions, specificity values were 0.0 under both evaluated conditions in this dataset. Therefore, these metrics should be interpreted as exploratory descriptors of assay behavior under the defined experimental conditions rather than as evidence of clinical diagnostic performance. The dNTP conditions were not included in this analysis because the number of evaluable positive results was limited (Table 4).
Performance metrics were calculated using predefined Ct-based operational criteria for internal comparative analysis and should not be interpreted as externally validated diagnostic accuracy estimates.
Since the first report of the SARS-CoV-2 and the onset of the pandemic, public health strategies have emphasized the need for rapid, cost-effective, and accurate diagnostic tools capable of processing large numbers of samples. Numerous RT-qPCR-based diagnostic systems have been developed targeting SARS-CoV-2 marker genes (Ruhan et al., 2020; Nalla et al., 2020). However, similar to the Berlin protocol (Corman et al., 2020), several of these approaches require sequential reactions, which may prolong diagnostic workflows and limit efficiency.
To our knowledge, this study represents one of the first approaches to evaluate the use of three gene targets both independently and in multiplex RT-qPCR reactions for SARS-CoV-2 detection. This strategy combined primers and probes from the E gene (Berlin protocol), the N gene (CDC protocol), and the ORF8 gene, together with reagents designed to reduce RNA secondary structures and account for nucleotide composition variability. These modifications were associated with changes in Ct values under the evaluated experimental conditions.
High-Performance Computing (HPC) analyses enabled large-scale SARS-CoV-2 genome alignment and database curation using publicly available sequences from GISAID and GenBank up to December 31, 2020 (Zhu et al., 2020). This approach supported primer and probe design as well as in silico evaluation. However, the compiled database may not fully represent the global distribution of viral variants, as sequencing capacity varies across regions.
The use of a global genomic dataset also enabled the prediction of RNA and cDNA secondary structures associated with amplified regions, informing primer and probe design. Additionally, this approach supported the formulation of denaturing solutions and nucleotide compositions that were associated with changes in Ct values under the evaluated conditions (Kovarova & Draber, 2000). These factors may contribute to differences in RT-qPCR assay performance within the experimental framework of this study.
The reagents and methods evaluated showed Ct values comparable to those reported in previous studies (Arakawa et al., 2024; Aranha et al., 2021; Chen et al., 2022). However, the proposed approach relies on the use of multiple gene targets and internal controls to ensure detection accuracy. Furthermore, the high mutation rate of SARS-CoV-2 may affect assay performance over time, highlighting the need for continuous updates of genomic databases.
Based on the results obtained, multiplex RT-qPCR represents a potential approach to address some of the limitations described. However, no consistent differences in Ct values were observed across all samples when compared with commercial methods. Additionally, the implementation of multiplex assays requires multiple fluorescent dyes and specialized equipment, which may increase operational costs. Despite these considerations, combining multiple gene targets may increase the likelihood of detecting a signal under specific conditions. The modular nature of the proposed approach allows flexibility in selecting gene targets according to diagnostic requirements.
Another advantage of multiplex RT-qPCR is the possibility of analyzing multiple samples from a single patient (Grobe et al., 2021). Although preliminary studies suggest that sampling at different time points may improve detection consistency, this approach requires additional clinical monitoring to optimize its implementation.
Taken together, these findings suggest that integrating HPC-based design, primer and probe optimization, and RT-qPCR protocols may support improvements in SARS-CoV-2 detection workflows under the evaluated conditions.
This study has several limitations. The sample size was constrained by logistical factors and low participation rates during the pandemic, including reluctance to undergo additional sampling procedures (McElfish et al., 2021), as well as compliance with legal and ethical requirements. Consequently, only participants who provided informed consent were included.
Additionally, sample classification was based on Ct threshold criteria without independent external validation. Therefore, the reported performance metrics reflect comparative assay behavior under the evaluated conditions rather than absolute diagnostic accuracy.
These limitations should be considered when interpreting the results. Further studies incorporating larger datasets and additional validation approaches are required to assess the broader applicability of the proposed methodology. In addition, clinical characterization of participants, including symptom severity and extent of infection, was not available for analysis because this information was protected under the informed consent framework and applicable legal restrictions.
The results of this study suggest that the incorporation of denaturing solutions and the adjustment of nucleotide proportions may influence RT-qPCR performance under the evaluated experimental conditions. These effects were observed in both single and multiplex assays, although variability was detected across samples.
The integration of computational approaches, including HPC-based genome analysis, with primer and probe design provided a framework to evaluate the potential impact of RNA secondary structure and nucleotide composition on assay performance.
Given the exploratory nature of this study and the limited sample size, these findings should be interpreted within the context of the experimental design. Additional studies incorporating larger datasets and independent validation strategies will be necessary to determine the broader applicability of the proposed approach.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)