Keywords
GRN analysis, single-cell RNA-seq, single-cell ATAC-seq, Gene regulatory network, Transcription Factor
This article is included in the Genomics and Genetics gateway.
This article is included in the Bioinformatics gateway.
GRN analysis, single-cell RNA-seq, single-cell ATAC-seq, Gene regulatory network, Transcription Factor
scANANSE is now compatible with Seurat version 5. Furthermore the manuscript is improved based on the suggestions of the peer reviewers. The specific alterations are written in the response to the peer reviewer reports.
See the authors' detailed response to the review by Marouen Ben Guebila
See the authors' detailed response to the review by Kenji Kamimoto
Single-cell RNA-sequencing (scRNA-seq) and single-cell ATAC-sequencing (scATAC-seq), enable measurement of gene transcripts (Islam et al., 2014) and genome accessibility (Buenrostro et al., 2015) at single-cell resolution. By performing single-cell sequencing on complex biological tissues and systems, various types of cells present in the system can be identified. Furthermore, gradual changes during development and differentiation trajectories can be scrutinised. The transcriptome and accessible genome of various cell populations can be quantified, which is not obtainable using bulk analyses (Huang, 2009; Li and Clevers, 2010). Capturing heterogeneity is vital in studying complex tissues, or while studying gradual processes such as development and differentiation, in which not all cells develop at the same rate or follow the same trajectory (Welch, Hartemink and Prins, 2016).
One of the main drivers of differences in cellular identity and developmental processes are transcription factors (TFs). To regulate gene expression, many TFs bind the DNA directly on DNA binding motifs. These motifs are present within cis-regulatory elements (CREs), which are functionally categorised as promoters, enhancers, or insulators (Lambert et al., 2018; Chen and Pugh, 2021). These cis-regulatory elements (CREs) can be used to scan for binding motifs. However, motif enrichment does not take into account the target of CREs, the nearby genes. To better predict the impact and importance of TFs, modelling gene regulatory networks (GRNs) is preferable.
By combining (differential) gene expression, genome accessibility, and motif enrichment, with the nearby location of target genes, it is possible to generate a directed GRN. Software to predict GRNs have been actively developed since the emergence of next-generation sequencing (Mercatelli et al., 2020). The addition of genome accessibility data and incorporation of long-range CREs is a successful method to model directed-GRNs (Xu et al., 2021; González-Blas et al., 2022; Kamal et al., 2022). Since both scRNA-seq and scATAC-seq are available, performing directed GRN analysis can now be applied to single-cell datasets.
There are multiple single-cell-based GRN tools available, capable of combining scRNA-seq and scATAC-seq data (Kamimoto, Hoffmann and Morris, 2020; Fleck et al., 2021; González-Blas et al., 2022; Kartha et al., 2022). However, since single-cell data contains relative low genome and/or transcriptome coverage per cell compared to bulk, one of the main challenges these tools face is using this sparse data. Furthermore, since these tools are specifically designed for single-cell data, making comparisons of their results with available bulk datasets is challenging.
In contrast, using single-cell data from robust clusters as pseudo-bulk can be used as input for many GRN tools available. To identify key TFs using GRN approaches, we previously developed the GRN analysis software ANANSE (Xu et al., 2021). ANANSE has multiple advantages: it incorporates CRE signal in 100kb windows, contains extensive TF binding models trained on the REMAP database, and can analyse data on all vertebrate species and even on non-vertebrate species with some additional steps. Theoretically, ANANSE could be run on single-cell pseudo-bulk data; however, the steps involved in generating data per cluster and running all the needed pairwise comparisons are labour-intensive and non-intuitive, while they require extensive bioinformatic skills.
Here, to enable ANANSE single-cell cluster analysis, we have developed an analysis pipeline called single-cell ANANSE (scANANSE). This pipeline consists of newly developed packages to export data from single-cell objects, either Seurat objects using the R implementation (AnanseSeurat), or from Scanpy objects using the Python implementation (AnanseScanpy). Next, an automated snakemake pipeline of ANANSE facilitates the GRN modelling. In parallel, it integrates motif enrichment analysis using GimmeMotifs (van Heeringen and Veenstra, 2011; Bruse and Heeringen, 2018). This addition is used to identify TFs with repressive properties, which are generally not properly predicted by ANANSE. Lastly, TFs influence score and motif enrichment results can be imported back into the single-cell object for downstream analysis and visualisation.
The performance of the scANANSE pipeline is demonstrated on a publicly available PBMC multi-omics dataset, as an example workflow including the installation of all software needed to run the analysis. In this PBMC case study, scANANSE uncovered many well-known activating TFs within the hematopoietic lineages. Including CEBPD and SPI1 in monocytes, EBF1 and MEF2C in B-cells, and STAT4 and LEF1 in T-cells. In addition, motif enrichment and expression correlation identify both the well known repressors PAX5 and STAT6 within B-cells.
The scANANSE pipeline consists of two components: a package to export and import data from and towards single-cell objects, and a snakemake implementation of ANANSE called anansnake (Figure 1). Crucial steps before running scANANSE are pre-processing, quality control, and clustering of single-cell data. For these steps, a large number of well-described workflows are available (Zappia and Oshlack, 2018; Luecken and Theis, 2019; Baek and Lee, 2020).
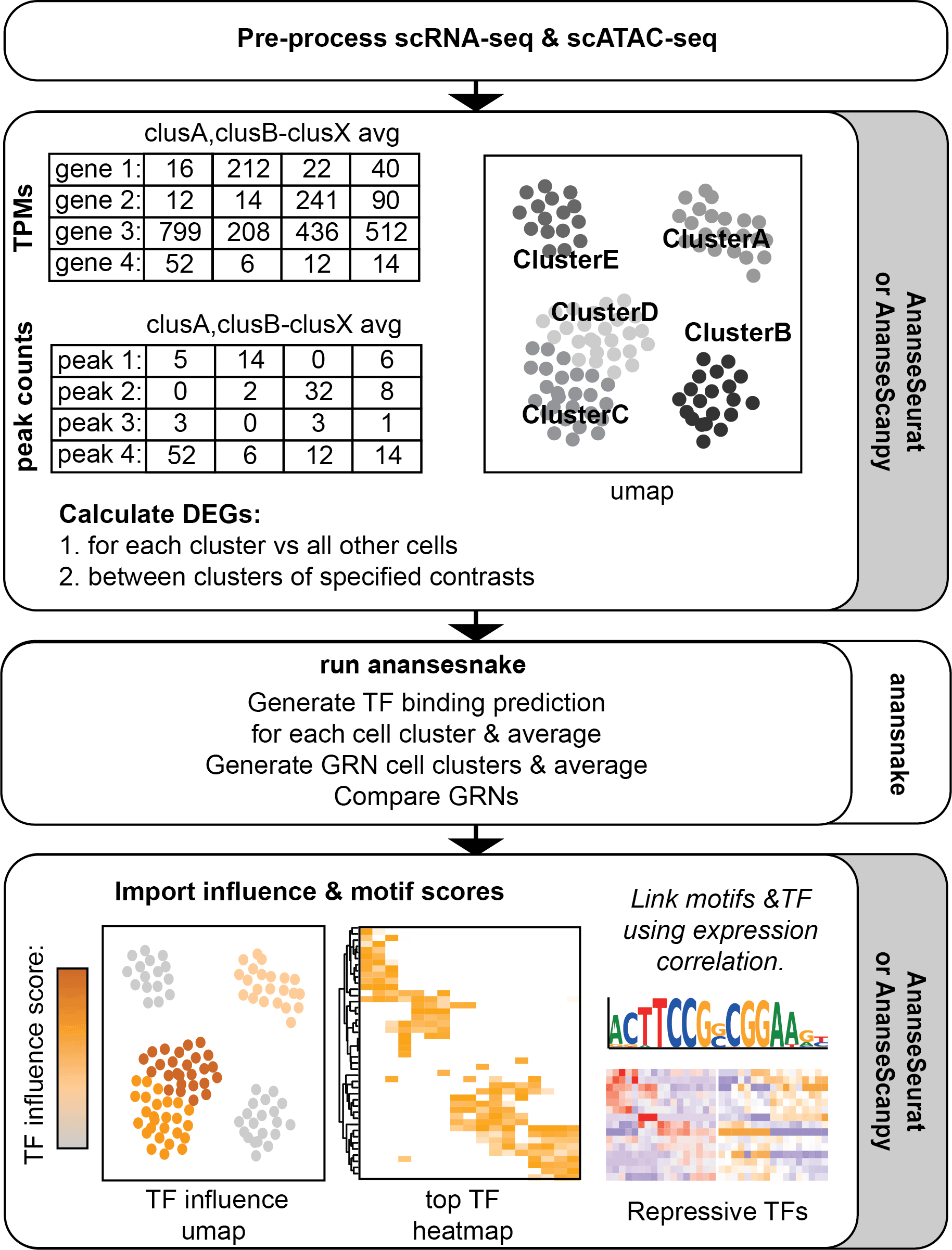
After pre-processing and clustering, data is exported using either AnanseSeurat or AnanseScanpy. Next, Anansnake automatically runs ANANSE after which the influence scores and motif enrichment results with AnanseSeurat or AnanseScanpy are imported. In parallel, Anansnake runs motif enrichment analysis using gimme maelstrom, and the motif results are imported and linked to the highest correlating TFs using the single-cell object scRNA-seq data.
scANANSE exports data from the single-cell object of choice. Transcripts Per Million (TPM), Differential Expressed Genes (DEGs) and peak counts need to be calculated based on the single-cell objects supplied. For Seurat objects (Hao et al., 2021) in the programming language R (R Core Team, 2021), the R package “AnanseSeurat” was developed to perform these steps. While for Scanpy objects (Wolf, Angerer and Theis, 2018) in the programming language Python (Van Rossum and Drake, 2009), the Python package “AnanseScanpy” was developed.
The TPM counts, DEGs, and ATAC peak counts can be exported from one single-cell object containing both the scRNA-seq data and scATAC-seq data, or from two separate single-cell objects. In the case of two single-cell objects, these objects need to share their cluster names, e.g. by transferring anchors between separate scRNA-seq and scATAC-seq datasets (Stuart et al., 2019). As such, scRNA-seq and scATAC-seq data from multiple studies or experiments can be combined and used as input.
By default, scANANSE compares each cluster to a GRN built from the average expression and gene accessibility of all clusters. This average network is used as a common comparison to compare all clusters. These comparisons result in an average GRN ‘TF-influence’ score. This score quantifies the importance of a TF driving the differences between a specific cluster and the average of all other cell clusters. In this way, the TF influence score can be compared across multiple clusters. In addition to this general approach, more detailed direct cluster-to-cluster GRN analyses are possible.
One downside of the GRN modelling of ANANSE is the lack of prediction of repressive TFs. This is based on underlying methods of ANANSE (Xu et al., 2021). To counteract this blind spot of the algorithm, motif enrichment with GimmeMotifs is performed in the scANANSE pipeline. It not only performs motif enrichment but is combined with a correlation of motif-z-scores and TF expression across clusters within the single-cell object. This addition enables the ability to predict repressive TFs.
Finally, both AnanseSeurat and AnanseScanpy can be used to import the TF influence and motif enrichment scores back into your single-cell object for further visualisation and analysis. All the source code and the conda environment YAML files used to generate the results presented in this article are available in Github and Zenodo (Arts et al., 2022).
The multi-omics dataset generated on human Peripheral blood mononuclear cells (PBMCs) publicly provided by 10× (PBMC from a Healthy Donor (v1, 150×150) Single Cell Multiome ATAC + Gene Expression Dataset by Cell Ranger ARC 2.0.0, 10× Genomics, 2022, December 20) is used as a case study. The scANANSE pipeline can also handle separate scRNA-seq and scATAC-seq objects with identical cluster names. However, within this example, scRNA-seq and scATAC-seq are part of the same single-cell object.
The package management system Conda is installed with two environments: anansnake and scANANSE. The following folder structure is used:
1a. Create folders
mkdir -p scANANSE/analysis mkdir -p scANANSE/data
1b. Install Conda
The operating system and computing environment are set up as listed in the minimal system requirements. Next, Conda is installed.
# Install Conda wget https://repo.anaconda.com/miniconda/Miniconda3-py38_4.12.0-Linux-x86_64.sh bash Miniconda3-py38_4.12.0-Linux-x86_64.sh rm Miniconda3-py38_4.12.0-Linux-x86_64.sh # Configure Conda conda config --add channels bioconda conda config --add channels conda-forge conda config --set channel_priority strict conda install mamba -y
1c. Install the anansnake Conda environment
mamba create -n anansnake anansnake
1d. Install the R Conda environment
wget https://raw.githubusercontent.com/JGASmits/AnanseSeurat/main/inst/scANANSE.yml mamba env create -f scANANSE.yml
1e. Install hg38
The location where Genomepy installs genomes is set using the -g flag. Since UCSC has three annotations for hg38, the version with HGNC gene names is selected, using --UCSC-annotation. scANANSE requires HGNC gene names to run.
conda activate anansnake genomepy install hg38 -g scANANSE/data --UCSC-annotation refGene
1f. Install AnanseSeurat and R packages
There are code blocks equivalent for exporting and visualising the data in python using Ananscanpy. See the extended data file “AnanseScanpy_equivalent.pdf” in the extended data (Arts et al., 2022) for these same steps but in Python. If RStudio needs to be installed on your system, see “install_Rstudio.pdf” in the extended data on Zenodo (Arts et al., 2022).
conda activate scANANSE rstudio
From R (studio):
install.packages("AnanseSeurat")
remotes::install_github("mojaveazure/seurat-disk", upgrade = "never")In this example we use data from 10x pre-processed by a vignette from Signac (2022). This dataset comes with a vignette performing default quality control, clustering, and annotation from the PBMC atlas from Hao et al. (2021). Proper quality control and clustering are vital for all single-cell analyses for these topics, however, there already exist some excellent reviews about these topics (Zappia and Oshlack, 2018; Luecken and Theis, 2019; Baek and Lee, 2020).
2a. Download the raw data (optional)
cd scANANSE/data wget https://zenodo.org/record/7575107/files/pbmc_granulocyte_sorted_10k_filtered_feature_bc_matrix.h5 wget https://zenodo.org/record/7575107/files/pbmc_granulocyte_sorted_10k_atac_fragments.tsv.gz wget https://zenodo.org/record/7575107/files/pbmc_granulocyte_sorted_10k_atac_fragments.tsv.gz.tbi wget https://zenodo.org/record/7575107/files/pbmc_multimodal.h5seurat cd ../..
2b. Pre-process single-cell data (optional)
An R Markdown file with all subsequent steps in R, including the pre-processing is available and can be downloaded.
wget https://raw.githubusercontent.com/JGASmits/AnanseSeurat/main/inst/scANANSE.Rmd -O scANANSE/scANANSE.Rmd
The pre-processing analysis follows the Signac multi-omics vignette (‘Signac’, 2022)
The QC steps can be skipped by downloading the processed Rds file.
wget https://zenodo.org/record/7575107/files/preprocessed_PBMC.Rds
Alternatively, the processed h5ad objects for AnanseScanpy can be downloaded.
wget https://zenodo.org/record/7575107/files/rna_PBMC.h5ad -O scANANSE/rna_PBMC.h5ad wget https://zenodo.org/record/7575107/files/atac_PBMC.h5ad -O scANANSE/atac_PBMC.h5ad
3a. Export cluster CPM, ATAC peak counts, and RNA-seq Counts
For the ATAC-seq data, a matrix containing the counts per peak per cluster is generated. For RNA-seq, CPM equivalent values are needed. Since the data is UMI normalised, CPM is already equivalent to regular depth normalised data (Phipson, Zappia and Oshlack, 2017). By default, scANANSE compares all clusters to a network based on the average values of all clusters. Additional comparisons can be specified, in this case, B-naive and B-memory cells were also specified to compare directly to each other.
conda activate scANANSE rstudio
# Load the required R libraries library(Seurat) library(SeuratDisk) library(stringr) library(ComplexHeatmap) library(circlize) library(ggplot2) library(AnanseSeurat) library(SeuratDisk) # Load pre-processed seurat object RDS file rds_file <- './scANANSE/preprocessed_PBMC.Rds' pbmc <- readRDS(rds_file) export_CPM_scANANSE(pbmc, in_cells <- 25, output_dir ='./scANANSE/analysis', cluster_id = 'predicted.id', RNA_count_assay = 'RNA') export_ATAC_scANANSE(pbmc, min_cells <- 25, output_dir ='./scANANSE/analysis', cluster_id = 'predicted.id', ATAC_peak_assay= 'peaks') # Specify additional contrasts: contrasts <- c('B-naive_B-memory', 'B-memory_B-naive') config_scANANSE(pbmc, min_cells <- 25, output_dir ='./scANANSE/analysis', cluster_id = 'predicted.id', additional_contrasts = contrasts) DEGS_scANANSE(pbmc, min_cells <- 25, output_dir ='./scANANSE/analysis', cluster_id = 'predicted.id', additional_contrasts = contrasts)
3b. File examples
Example of values and layout of the TPM.tsv file generated by the export_CPM_scANANSE() function.
| CD4-Naive | CD4-TCM | average | |
|---|---|---|---|
| MIR1302-2HG | 0 | 0 | 0 |
| FAM138A | 0 | 0 | 0 |
| OR4F5 | 0 | 0 | 0 |
| AL627309.1 | 236 | 1.007 | 1.118 |
Example of values and layout of the Peak_Counts.tsv file generated by the export_ATAC_scANANSE() function.
| CD4-Naive | CD4-TCM | Average | |
|---|---|---|---|
| chr1:10032-10322 | 5 | 22 | 5 |
| chr1:180709-181030 | 6 | 20 | 5 |
| chr1:181296-181600 | 10 | 12 | 5 |
| chr1:191304-191914 | 7 | 12 | 5 |
Next, snakemake is run from within a screen session. This takes approximately 3 hours per cluster plus 2 hours for motif enrichment analysis, but this is also highly dependent on computer speed. With less than 64 GB of RAM available, we recommend downscaling the core number to a maximum of 6 cores. With more RAM and more cores available the core count should be increased to reduce analysis time.
Additionally, it is possible to add extra samples and/or networks to the anansnake run. This enables including other samples and other networks in your comparisons. When performing additional anansnake comparisons please go through the anansnake documentation in detail.
screen
conda activate anansnake
# Update the timestamps so snakemake doesn't try to regenerate the DEG files if you make changes to the config or sample file
for DEGfile in scANANSE/analysis/deseq2/*;do touch -m $DEGfile;done
anansnake \
--configfile scANANSE/analysis/config.yaml \
--resources mem_mb=48_000 --cores 125a. Import the ANANSE results
After running ANANSE with anansnake, the influence output is imported back into the single-cell object.
conda activate scANANSE rstudio
pbmc <- import_seurat_scANANSE( pbmc, cluster_id = 'predicted.id', anansnake_inf_dir = "./scANANSE/analysis/influence" ) # export the data per cluster from the single-cell object TF_influence <- per_cluster_df(pbmc, assay = 'influence', cluster_id = 'predicted.id') head(TF_influence)
Example of the influence data frame generated by per_cluster_df(assay = ‘influence’).
| B-intermediate | B-memory | B-naive | CD14-Mono | CD16-Mono | |
|---|---|---|---|---|---|
| EBF1 | 1 | 1 | 1 | 0 | 0 |
| SPIB | 0.9375 | 0.913043 | 0 | 0 | 0 |
| REL | 0.875 | 0.391304 | 0.947368 | 0.818182 | 0.943396 |
| TCF4 | 0.8125 | 0.695652 | 0.736842 | 0 | 0 |
| BACH2 | 0.75 | 0.391304 | 0.894737 | 0 | 0 |
5b. Top five influential TFs per cluster
Next, the top five TFs per cluster are identified from the influence table.
TF_influence$TF <- rownames(TF_influence) TF_long <- reshape2::melt(TF_influence, id.vars = 'TF') colnames(TF_long) <- c('TF','cluster', 'influence') TF_influence$TF <- NULL TF_long <- TF_long[order(TF_long$influence, decreasing = TRUE), ] # get the top n TFs per cluster topTF <- Reduce(rbind, by(TF_long, TF_long["cluster"], head, n = 5))# Top N highest TFs by cluster top_TFs <- unique(topTF$TF) TF_table <- topTF %>% dplyr::group_by(cluster) %>% dplyr::mutate('TopTFs' = paste0(TF, collapse = " ")) unique(TF_table[,c('cluster','TopTFs')])
Referenced TFs in the text are in bold and highlighted.
5c. Heatmap of most influential TFs
An overview of the top TF and their various influences in the various clusters is visualised by a heatmap. The column and row dendrogram are manually swapped where appropriate resulting in the final TF influence heatmap (see Figure 2).
col_fun = circlize::colorRamp2(c(0, 1), c("white", "orange")) mat <- as.matrix(TF_influence[rownames(TF_influence) %in% top_TFs,]) pdf('./scANANSE/analysis/ANANSE_Heatmap.pdf',width=16,height=8,paper='special') ComplexHeatmap::Heatmap(mat, col = col_fun) dev.off()
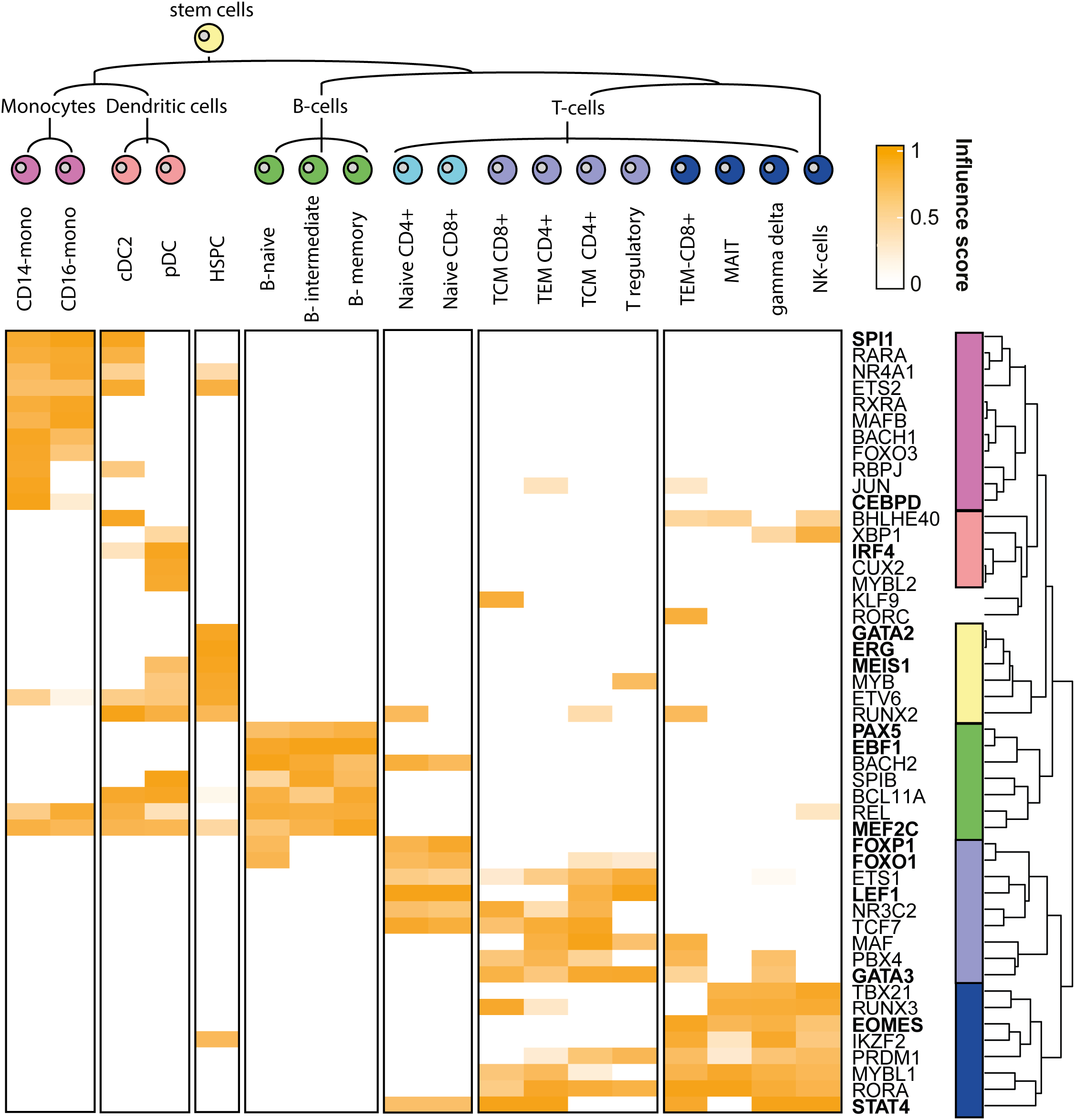
This heatmap depicts the influence scores of the top five highest influential TFs per cluster. Referenced TFs in the text are in bold.
By using scANANSE, a large number of well-known hematopoiesis hallmark TFs is identified (see Figure 2 and Table 5). This demonstrates the ability of scANANSE to identify important TFs from single-cell data. Some well-known examples are:
Monocytes TFs include, SPI1 which is well known to regulate human monocyte differentiation towards dendritic cells and is identified in both monocytes and dendritic cells (Rosa et al., 2007; Novershtern et al., 2011; Zhu et al., 2012). While the CEBP gene family, including the identified CEBPD, is vital for the transduction of B-cells into macrophages (Bussmann et al., 2009).
Dendritic cell TFs including IRF4 (Tamura et al., 2005) were identified as driving Interferon producing pDCs (Siegal et al., 1999).
Hematopoietic stem cell TFs include GATA2 (Menendez-Gonzalez et al., 2019, p. 2), ERG (Knudsen et al., 2015) and MEIS1 (Novershtern et al., 2011; Ariki et al., 2014). All these factors are all well-known regulators of hematopoietic stem cell identity.
B-cell TFs include EBF1 and MEF2C. Both are well-known to drive the B-cell lineage (Kong et al., 2016; Bullerwell et al., 2021, p. 1), while PAX5 is another well-known B-cell fate driving factor (Enver, 1999, p. 5; Medvedovic et al., 2011, p. 5). In particular, PAX5 is an intriguing finding since it is not only well known to promote B-cell genes, but also to repress non-B-cell lineage genes (Boller and Grosschedl, 2014). This repressive property is however not included in the ANANSE analysis. And its prediction is likely attributed to the smaller effect of gene activation PAX6 has on specific target genes.
T-cell TFs include both GATA3 and LEF1, which are crucial for specifying the T-cell fate (Novershtern et al., 2011). Furthermore, more specific to naive T-cells, FOXO1 (Kerdiles et al., 2009, p. 1) and FOXP1 (Feng et al., 2010) are known to maintain naive T-cell quiescence.
Differentiated T-cell TFs include the well-known STAT4 (Novershtern et al., 2011; Suarez-Ramirez et al., 2014), and for both CD8+ T-cells and NK cells the well-known TF EOMES (Shimizu et al., 2019) are identified.
5d. Visualise TF expression and influence on a UMAP
The presence of the influence scores enabled clear visualisation of the influence and expression of specific TFs across the dataset. As an example, three TFs are visualised with a wide variety of influence and expression across clusters (see Figure 3).
highlight_TF1 <- c('STAT4','LEF1','MEF2C') Annotated_plot <- DimPlot(pbmc, label = T, repel = TRUE, reduction = "umap")+ NoLegend() DefaultAssay(object = pbmc) <- "RNA" plot_expression <- FeaturePlot(pbmc, features = highlight_TF1, ncol = 1) DefaultAssay(object = pbmc) <- "influence" plot_ANANSE <- FeaturePlot(pbmc, ncol = 1, features = highlight_TF1, cols = c("darkgrey", "#fc8d59")) pdf('./scANANSE/analysis/ANANSE_highlight.pdf',width=10,height=10,paper='special') print(Annotated_plot) print(plot_expression|plot_ANANSE) dev.off()
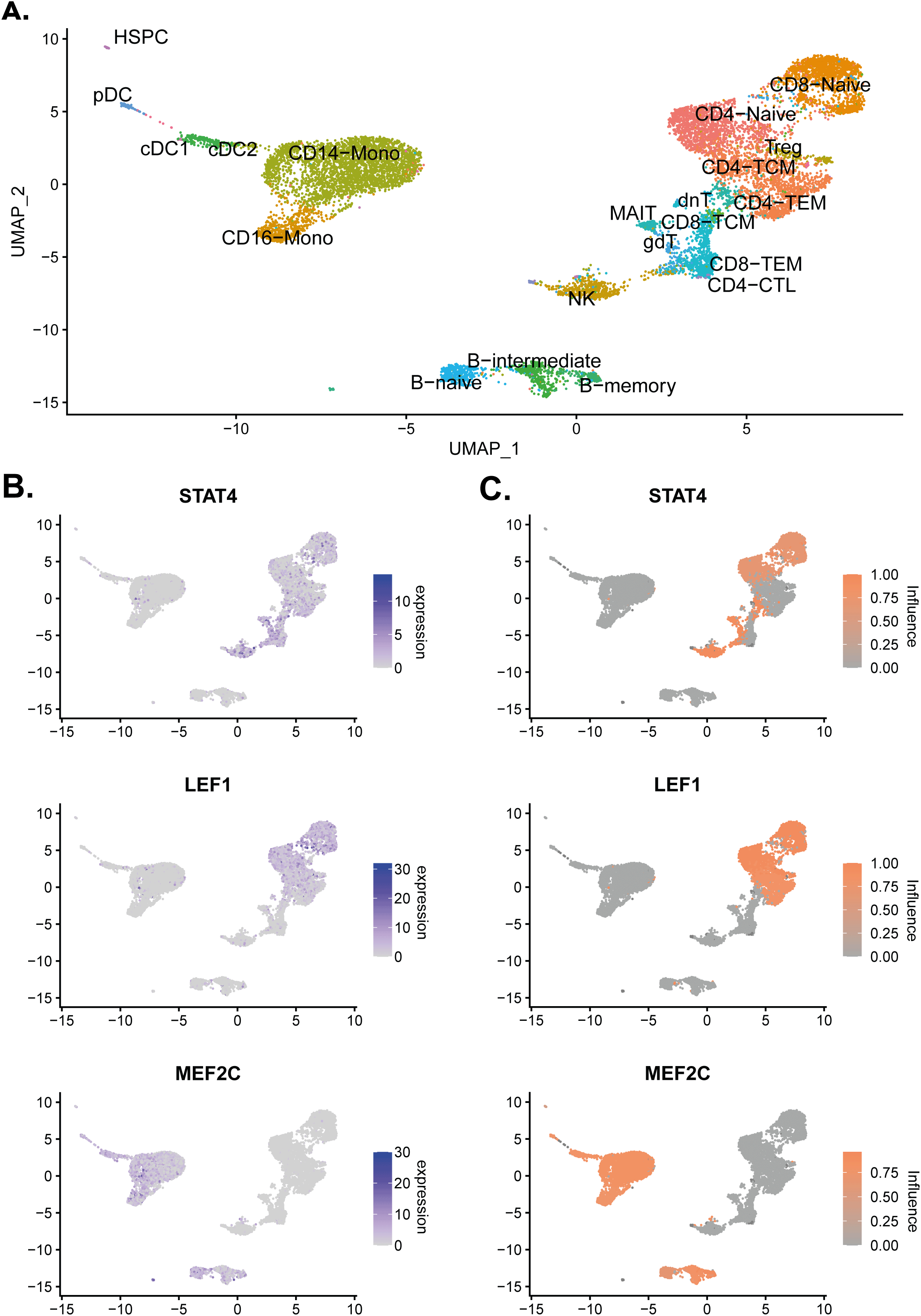
(A) UMAP of the PBMC single-cell object with the cell identities labelled. (B) Normalised expression values of STAT4, LEF1, and MEF2C on the single-cell object. (C) Influence scores of STAT4, LEF1, and MEF2C on the single-cell object.
Although all B-cell clusters were relatively similar when compared to the average network, it is possible to directly compare both clusters. This uncovers TFs driving more subtle differences between the cell types. This direct cluster-to-cluster comparison is performed by adding the two clusters in part 3 as an additional contrast.
When comparing Naive B-cells and Memory B-cells, FOXP1 and BACH2 were identified as important factors driving Memory B-cell maturation compared to naive B-cells. This is in line with previous publications (Itoh-Nakadai et al., 2014; Patzelt et al., 2018). Furthermore, EBF1 and SPIB were identified as driving Naive B-cells, this is also in line with previous research (Schmidlin et al., 2008; Györy et al., 2012). Thus, these results illustrate the possibility of running comparisons on similar clusters within single-cell datasets to further identify TF networks that define cell types (Figure 4).
MemoryInfluence <- read.table( './scANANSE/analysis/influence/anansesnake_B-memory_B-naive.tsv', header = T) NaiveInfluence <- read.table( './scANANSE/analysis/influence/anansesnake_B-naive_B-memory.tsv', header = T) NaiveInfluence$factor_fc <- NaiveInfluence$factor_fc* -1 B_comparison <- rbind(NaiveInfluence,MemoryInfluence) ggplot(B_comparison, aes(factor_fc,influence_score)) + geom_point(aes(size = direct_targets, colour = influence_score)) + xlim(-2,2)+ geom_text( aes( label=ifelse(factor_fc > 0.26|factor_fc < -0.5,as.character(factor),""), hjust = 0.5, vjust = 2 ))
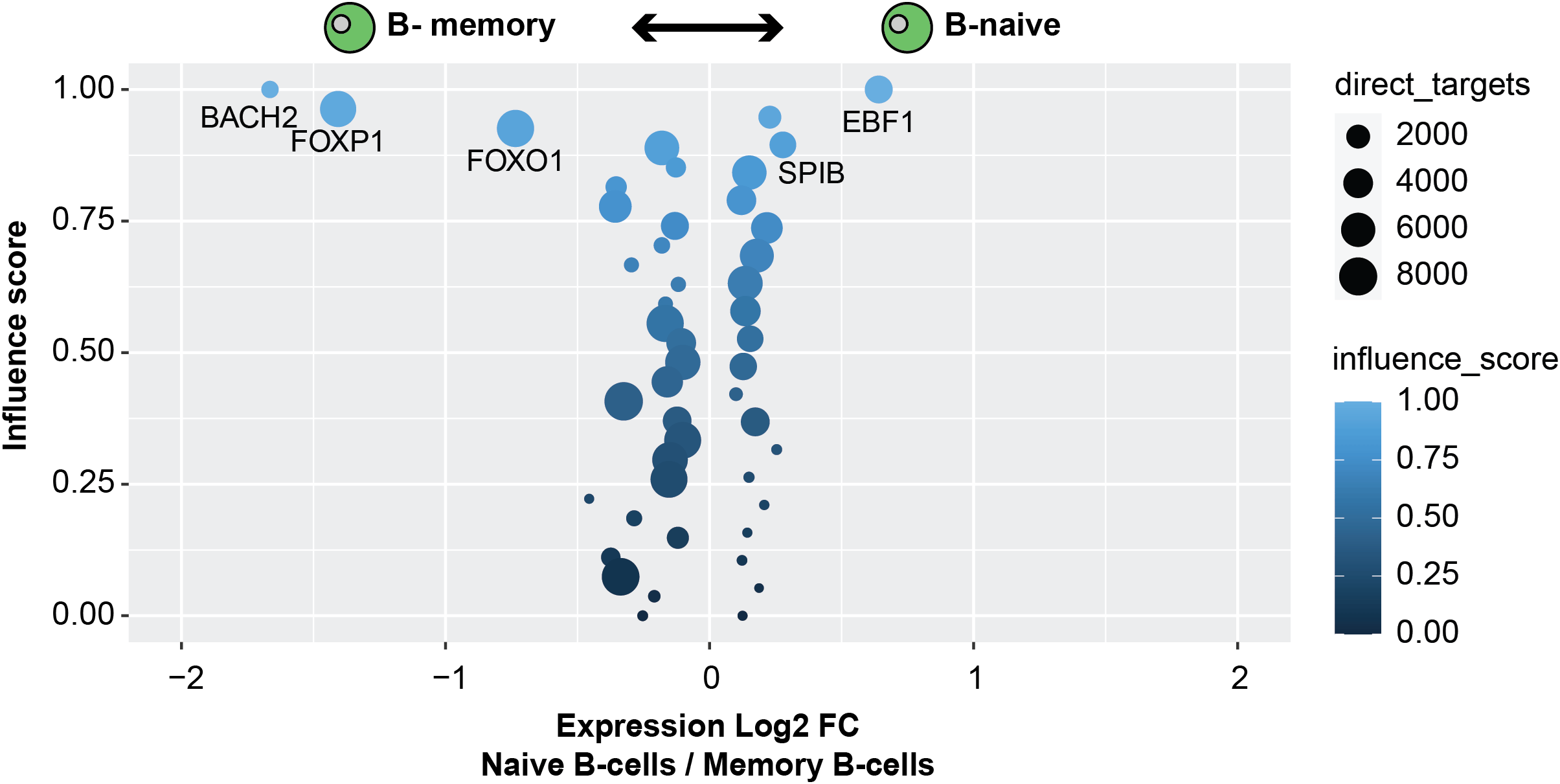
The TF influence scores of TFs comparing Naive B-cells and Memory B-cells; higher influence of factors with negative fold changes are more important within memory B-cells; higher influence of factors with positive fold changes are more important in Memory B-cells. Circle size correlates with the number of direct target genes. Gene expression log2 fold change between Naive B-cells and memory B-cells on the X axis.
The original ANANSE method was not developed to reliably predict repressive factors (Xu et al., 2021). Instead motif enrichment can be used for identifying motifs with reduced accessibility, however due to the lack of a one-on-one link of motifs and TFs, and the difference of these interactions between tissues, it is tricky to reliably link motifs with their most relevant factors in the cell type of interest.
However, with single-cell cluster data, it is possible to link motifs and TFs based on motif and expression correlation across multiple clusters. This approach does enable scANANSE to identify potential repressive factors. It is however a step down from the GRN modelling approach, but for identifying potential repressive factors it is an easy step to incorporate, which we therefore choose to include.
We will first incorporate the enrichment result after running anansesnake.
Import motif enrichment scores
pbmc <- import_seurat_maelstrom(pbmc, cluster_id = 'predicted.id', maelstrom_file = './scANANSE/analysis/maelstrom/final.out.txt') # export the data per cluster from the single-cell object motif_scores <- per_cluster_df(pbmc, assay = 'maelstrom', cluster_id = 'predicted.id') head(motif_scores)
Example of the Motif score data frame generated by per_cluster_df(assay = ‘maelstrom’).
Link TFs to motifs based on their correlation coefficient
The enriched motifs are linked to TFs based on the non-redundant motif-TF database generated by GimmeMotifs. A correlation score is calculated between the motif-z-scores and TF expression values. When multiple TFs map to the same motif of interest, the TF with the highest absolute correlation is linked to this motif. After linking all motifs, one TF can be linked to multiple motifs. In that case, there are multiple options for selecting the most relevant motif.
First of all, it is possible to take the mean motif score, secondly by selecting the motif with the most variable signal, or thirdly by selecting the motif with the highest absolute correlation between enrichment and expression. Here we use the motifs with the highest correlation to the expression.
Finally, two assays are added to the single-cell object, one consisting of a positive correlation with linked motifs, which indicates a TF promoting genome accessibility, and one assay consisting of a negative correlation with linked motifs, which indicates TFs repressing genome accessibility. A TF can be present in both assays when it is linked both with a motif with a positive correlation and a motif with a negative correlation.
pbmc <- Maelstrom_Motif2TF(pbmc, cluster_id = 'predicted.id', maelstrom_dir = './scANANSE/analysis/maelstrom/', RNA_expression_assay = "SCT", expr_tresh = 10, cor_tresh = 0.3, combine_motifs = 'max_cor')
Visualise TF expression and motif enrichment
Next, the top TFs of with a negative correlation were visualised as a heatmap (Figure 5A).
col_fun <- circlize::colorRamp2(c(-5,0,5), c('#998ec3','white','#f1a340')) col_fun_cor <- circlize::colorRamp2(c(-1,0,1), c('#7b3294','#f7f7f7','#008837')) for (regtype in c('TFcor','MotifTFanticor')){ top_TFs <- head(pbmc@assays[[regtype]][[]],15) mat <- per_cluster_df(pbmc, assay = regtype, cluster_id = 'predicted.id') mat <- as.matrix(mat[rownames(mat) %in% rownames(top_TFs),]) #get TF expression matrix exp_mat <- AverageExpression(pbmc,assay='SCT', slot = 'data', features = rownames(top_TFs), group.by = 'predicted.id')[[1]] exp_mat <- exp_mat[,colnames(exp_mat)] exp_mat <- as.matrix(t(scale(t(exp_mat)))) #get correlation score row_ha = rowAnnotation(correlation = top_TFs$cor, col = list(correlation = col_fun_cor)) print(Heatmap(exp_mat[,cluster_order], cluster_columns = F) + Heatmap(mat[,cluster_order], col = col_fun, cluster_columns = F, right_annotation = row_ha)) }
This identified multiple repressive hallmark TFs (Figure 5A). Examples and well known important repressors driving hematopoiesis include PAX5 (Souabni et al., 2002, p. 1), STAT6 (Czimmerer et al., 2018), ID2 (Ji et al., 2008), and PRDM1 (Chan et al., 2009, p. 1; Nadeau and Martins, 2022).
TF_list <- c('PAX5','STAT6') Factor_Motif_Plot(pbmc, TF_list, assay_maelstrom = 'MotifTFanticor', logo_dir = './scANANSE/analysis/maelstrom/logos/')
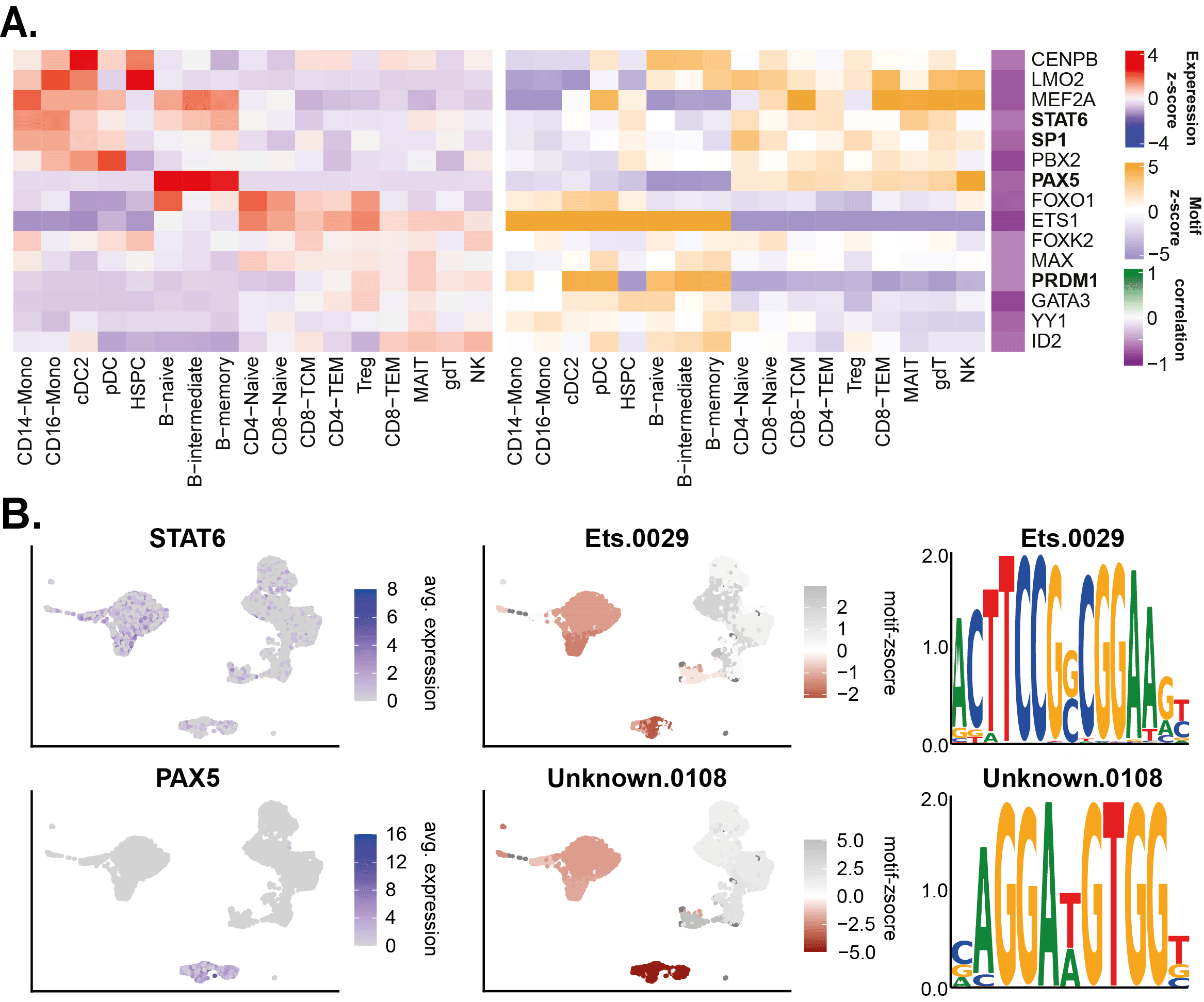
(A) Heatmap of top negatively correlating motifs & TFs. (B) UMAP example of anti-correlation factors PAX5 and STAT6.
Here we demonstrate that scANANSE is able to decipher the GRNs driving the identity of single-cell clusters. This enables the identification of TFs that drive the cellular identity of single-cell clusters of scRNA-seq and scATAC-seq datasets.
Currently, there are multiple other tools available and under development for performing GRN analysis using a combination of scRNA-seq and scATAC-seq data. Examples include software such as SCENIC+ (González-Blas et al., 2022), Pando (Fleck et al., 2021), CellOracle (Kamimoto, Hoffmann and Morris, 2020) and FigR (Kartha et al., 2022). These tools have the advantage and the challenge of calculating GRNs using individual cells. While they are not relying on clustering before GRN analysis, these tools struggle at identifying low expressed target genes and TFs since individual cells have low transcriptome coverage. Comparing and benchmarking all these single-cell GRN tools is beyond the scope of this paper, but would be an exciting addition to the field in the future.
scANANSE has some clear advantages. First of all, it has the ability to analyse single-cell data generated from all vertebrate genomes. When working with non-vertebrate data, extra steps for identifying homologous genes across phyla are required before running scANANSE. For more information on that topic, see the ANANSE documentation on the motif database. This flexibility enables GRN analysis on single-cell data from a high variety of organisms. Furthermore, due to the pseudo-bulk approach, it is possible to compare single-cell cluster gene regulatory networks against networks generated from traditional bulk sequencing data. Although the amount of publicly accessible single-cell datasets is growing, there is an even larger amount of bulk sequencing datasets available. Moreover, the possibility and flexibility of comparing GRNs from multiple sources is another advantage of scANANSE, extra care and validation is still needed when using networks from different data sources.
scANANSE makes a few assumptions that are important to note regarding the average network comparison. Using the average network as the background comparison against each cluster-specific network enables the identification of TFs driving each specific cluster. In the case of small cluster numbers, this approach is however limiting the reliability and the number of factors identified since the average network contains accessibility data from all clusters including the cluster being compared. In cases with low cluster numbers, it is therefore recommended to run scANANSE including pairwise comparisons between all the clusters.
Another limitation of the GRN modelling of ANANSE is its inability to predict repressive TFs, or factors with context-dependent and/or repressive properties (Krishnakumar et al., 2016; Pang and Snyder, 2020). While deciphering molecular mechanisms, the inclusion of repressive factors and factors with context-dependent purpose is highly useful (Gaston and Jayaraman, 2003; Bauer, Buske and Bailey, 2010; Arnold et al., 2013). ANANSE however uses a rank mean approach which assumes all TF target gene relations are activating, while furthermore requiring a TF to be higher expressed. These assumptions are not always applicable to TFs with repressive or context-dependent functions (Xu et al., 2021). To alleviate some of this limitation, we have integrated motif enrichment analysis from the GimmeMotifs toolkit. Combining the motif z-score with a correlation of TF expression provides a straightforward tool to link motifs to the most relevant TFs which can be repressive. However, this approach does not take into account the potential combinatorial function of TFs (Zeitlinger, 2020) and/or missing interactions in the TF to motif database.
With scANANSE, we have implemented a robust and capable toolkit to identify key TFs important for driving cellular identity and differentiation in single-cell data. It relies on solid pseudo-bulk signals and proven bulk-GRN approaches to identify the TFs of interest.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)