Keywords
Epistasis, genomic prediction, machine learning, rhizomania, resistance breeding, Beet necrotic yellow vein virus, variable importance
This article is included in the Bioinformatics gateway.
This article is included in the Genomics and Genetics gateway.
This article is included in the Plant Computational and Quantitative Genomics collection.
Epistasis, genomic prediction, machine learning, rhizomania, resistance breeding, Beet necrotic yellow vein virus, variable importance
Sugar beet (Beta vulgaris L.) is an important crop to secure production of white sugar, especially in industrialised countries.1 In general, sugar beet makes up 20% of the sugar production in the world.2 Next to high sugar yield, resistance to diseases, of which rhizomania is the most significant, is the main goal of sugar beet breeding.3
Rhizomania is caused by the Beet necrotic yellow vein virus (BNYVV)4 and is transmitted via the fungus Polymyxa betae Keskin.5,6 Severe infection with rhizomania can cause a reduced sugar yield of up to 90%.7 Moreover, Abe and Tamada (1986) have shown that rhizomania can persist in resting spores of P. betae for over fifteen years, making a decontamination through an enlarged crop rotation nearly impossible.8 Furthermore, there is no pesticide available for plant protection,9 leaving resistance breeding as the only defence strategy at the moment.10
Since the first observation of rhizomania in 1951 in northern Italy, the disease has spread globally and occurs nowadays in all major sugar beet growing regions in the world.11 While strain groups of BNYVV with four ribonucleic acid (RNA) strands are spread globally,12 BNYVV strains with five RNA strands have been found in certain regions in France,13 Japan,14 the UK,15 Kazakhstan,16 and Turkey.7 Comparisons between their pathogenicities revealed that pathotypes of BNYVV with five RNA strands showed significantly higher levels of infection in partially resistant sugar beet varieties than pathotypes with four RNA strands.17
The first breeding projects against rhizomania started in 197018 and resulted in the publication of three resistance genes in 1987 called “Rizor”,19 “Holly”,20 and “WB42”.20 Nevertheless, further analyses of the resistance genes “Rizor” and “Holly” indicated that these are probably the same gene, henceforth called “Rz1”.21 The resistance gene “WB42” which is also often referred to as “Rz2”, however, was assumed to be a further resistance gene independent from Rz1 with an approximate distance of 20 cM between Rz1 and Rz2.22 Recent studies could not only prove the existence of the Rz2 resistance gene in wild relatives of sugar beet but also found a stop codon in Rz2 in susceptible genotypes which was not present in resistant genotypes.23 In the following years, three further resistance genes were published called “Rz3”,24 “Rz4”,25 and “Rz5”.26
Although five resistance genes against rhizomania have been published, doubts have been raised on whether all resistance genes are in fact separate genes or rather alleles of the same genes. All five resistance genes were located on chromosome three18,25,26 where mainly two clusters emerged.27 McGrann et al. (2009) assumed that the resistance against rhizomania may be mainly explained by two loci, with the first locus being represented by Rz1, Rz4, and Rz5, and the second locus being represented by Rz2 and Rz3.12 Although only two resistance clusters against rhizomania are known, it is assumed that rhizomania resistance is a quantitative trait caused by multiple loci with different effects, which have not yet been identified.18
In general, it has also been postulated that asymmetric variation in quantitative traits can be caused by epistasis.28 Epistasis can be defined as non-additive interaction between genes.29 Theoretically, this interaction can be formed by more than two genes but the analysis of an interaction between more than two genes is challenging due to the computational complexity and the requirement to have large enough samples for each subgroup.30 Although it has been demanded to analyse epistasis in complex trait studies31,32 and epistasis has been analysed for a multitude of traits in sugar beet,33 such a study has not yet been conducted for rhizomania resistance to the best of our knowledge.
It is generally assumed that a plant’s resistance towards diseases is quantitative and caused by a complex network of multiple loci.34,35 In such cases, genomic prediction is a useful tool to predict an individual’s resistance towards the disease. Such studies have been performed, for example, in soy bean,36 barley,37 rapeseed,38 rice,39 wheat,40 and maize.41,42 Although rhizomania resistance is believed to be a complex trait caused by multiple loci, genomic prediction of rhizomania resistance has not yet been published. Here, we present the first study of genomic prediction of rhizomania resistance with 9,127 single nucleotide polymorphism (SNP) markers in a sugar beet population that is assumed to carry the known resistance genes (Rz1 and Rz2).
Our aim is to maximise the accuracy of genomic prediction of rhizomania resistance in sugar beet. Therefore, we performed genomic prediction using random forest with all available SNP markers. Moreover, we estimated the variable importance of each SNP and, subsequently, performed incremental feature selection to optimise the prediction model by only including an optimal set of SNPs. Furthermore, we used the SNP markers to create SNP pairs and performed genomic prediction with the SNP pairs instead of the single SNPs. Finally, we used the information about the variable importance of each single SNP to create SNP pairs with only the best SNP markers and selected the optimal set of SNPs for genomic prediction with SNP pairs. We provide an R script as well as the data from this trial to encourage researchers to perform feature selection with SNP pairs in their studies. The R script as well as the data from this trial have been published in version v1.0 at https://github.com/tmlange/IFS\_SNPpairs.git.43
The greenhouse test was performed with 156 genotypes. For each genotype, 15 plants were grown. Therefore, sugar beet lines were used which were created by crossing two sugar beet lines and self-pollinating the resulting hybrids two times. From the resulting seeds, 15 were chosen from each of the S2 plants as seeds for this trial and the genotype of the parent was assumed as the genotype of each of the seedlings. Thus, the parent was analysed using a SNP chip and the genomic data were used for the descendants. From the 156 genotypes that were used in this trial, 155 genotypes carried the resistance at the two known genes in homozygous form. Thus, it can be assumed that the descendants from these plants must carry the resistance in a homozygous form as well. One genotype, however, was susceptible at Rz2. Also here, it can be assumed that the descendants from this plant must be susceptible in a homozygous form.
Plants were grown for ten weeks in the greenhouse in soil infested with BNYVV, pathotype P. This variant of BNYVV contains five RNA strands44 and is, thus, assumed to be more aggressive than the variants with four RNA strands.17 After ten weeks, plants were removed from the soil and plant sap from lateral roots was extracted. Afterwards, the optical density (OD) value of each sample was measured using the double antibody sandwich enzyme-linked immunosorbent assay (DAS-ELISA). The OD values were measured after 60 minutes, 90 minutes, and 120 minutes using the Infinite F50® (Tecan Group AG, Männedorf, Switzerland) at a wavelength of 405 nm. Harvest, sample preparation and conduction of the DAS-ELISA test followed the protocol described in.45
Although DAS-ELISA is an often used tool to measure the concentration of BNYVV in samples from sugar beet,7,10,23 it does not directly measure the virus concentration in form of the OD values.46 To estimate the virus concentrations from the raw OD values as well as to reduce measurement errors for each 96-well plate, we transformed the non-normally distributed raw data to normally distributed data with an inverse logistic regression model.47 Therefore, a logistic regression model was derived using a serial dilution with 12 samples on each 96-well plate. The transformation followed the protocol in Ref. 47 with one adjustment: The ODs were not only measured after 90 minutes but also after 60 and 120 minutes. Thus, each sample had three OD measurements and three transformed measurements. As described in Ref. 47, it can be assumed that the transformed data are normally distributed. Thus, the mean of the transformed data has been calculated and used as response variable to reduce technical errors during measurement at each time point. A statistical model was fit to the data points in the serial dilution and, subsequently, OD values were transformed according to equation 1:
After the data set was transformed, the mean of the transformed data was calculated for all plants which were descendants of the same parent. Thus, if no plants died during the trial, the mean from 15 plants was calculated as the phenotypic data point for the corresponding genotype.
After transformation and calculating the mean of the transformed values, the SNPs were prepared. For this, SNPs with missing values were removed from the data set. Moreover, redundant SNPs were removed through linkage disequilibrium pruning. In this step, one of two SNPs that were correlated with more than an of was removed. This step should ensure that epistasis results were not confounded by linkage disequilibrium.32,48 Furthermore, SNPs with a major allele frequency of 0.95 or higher were removed. After the final step of SNP filtering, 9,127 SNPs were kept in the data set. Furthermore, since only one of the 155 genotypes was susceptible at Rz2, it can be assumed that the SNPs in high linkage disequilibrium with Rz2 must have been removed in the last step of filtering.
Finally, the remaining SNPs were recoded as 0 (homozygous major allele), 2 (homozygous minor allele), and 1 (heterozygous). This kind of coding was recommended for analysing genotypic and additive genetic models.49 All filtering steps and the recoding of the SNPs were performed using PLINK v1.90b6.10.50
After data were prepared, genomic prediction was performed using single SNPs. Therefore, the data set was divided randomly into 80% training data (125 data points) and 20% test data (31 data points). Following the experimental design in other studies that used feature selection,51 this process was repeated 10 times. Subsequently, genomic prediction was performed using random forest. To do so, the R package ranger, version 0.14.1 was used with default settings.52
Prediction accuracy was evaluated by prediction of the test data set with a prediction model that was derived using the training data set. Subsequently, the coefficient of determination () was used to compare the predicted values to the observed values in the test data set. The coefficient of determination is defined as the proportion of the explained variability of the total variability53 and was used in previous studies as measure for the prediction accuracy.54,55
To perform feature selection, the variable importance of each SNP was estimated using random forest in a first step. Therefore, the R package Boruta, version 7.0.0 was used.56 The R function Boruta from this package performs multiple random forest runs with the given input data and calculates multiple quantities as the resulting variable importance per input variable. This function was used in previous studies to assess the variable importance of SNPs.54,57,58 We chose the mean variable importance as estimate of the variable importance per SNP. We used a confidence level of , 2,000 trees and 200 as the maximum number of runs.
After the variable importance per SNP was estimated, feature selection was performed by carrying out genomic prediction with a random forest model that contained only the two SNPs with the highest variable importance. Subsequently, genomic prediction was performed using the three best SNPs, and so on. For each number of SNPs in the prediction model, the prediction accuracy as explained above was calculated. Since random forest can lead to different results in prediction accuracy even if the same data were used as training and test data set, prediction accuracy was estimated as the median prediction accuracy from ten repetitions. To prevent over-optimistic values for the prediction accuracy, variable importance was only estimated using the training data set.59 In this way, genomic prediction using feature selection can be compared to genomic prediction using all available SNPs.51,60
Performing the feature selection method as described above led to a variable importance per SNP as well as a prediction accuracy for each prediction model containing the best SNPs for each of the ten random splits of the data set. In a next step, the prediction accuracy for each number of SNPs was defined as the median from these ten repetitions. Subsequently, the optimal number of SNPs was defined as the number of SNPs that maximised the median prediction accuracy from the ten repetitions.
Besides genomic prediction with single SNPs, genomic prediction was also performed using SNP pairs. Since the genomic data set contained 9,127 SNPs after the data preparation and filtering, theoretically more than 41 million SNP pairs could be created out of these single SNPs. To reduce the resulting data set to a size that a computer can handle, PLINK’s epistasis test was performed using default settings. In this way, the interaction term of each two SNPs is tested for significance at a significance threshold of .61 The selection of SNP pairs was performed with each training data set individually to prevent bias during the selection process.
After the SNP pairs were selected using PLINK’s epistasis test, the genotype of each SNP pair was defined using an additive-additive interaction model. In this way, the genotype of each SNP pair was defined as the product of both single SNPs.49 Thus, if any of the single SNPs was homozygous for the major allele (coded as 0 in the single SNPs), the genotype of the SNP pair was defined as 0. This was the case for five of the nine possible genotypes of a SNP pair. The combination of two heterozygous single SNPs would lead to a 1 for the SNP pair, the combination of a heterozygous SNP with a SNP that provides a homozygous minor allele would be 2, and the combination of two SNPs that provide homozygous minor alleles would be 4. The recoding of single SNPs as well as the resulting genotype of the SNP pair is summarised in Table 1.
A and B represent major alleles and a and b represent minor alleles for SNP A and SNP B, respectively. The resulting SNP pair corresponds to the product of the single SNPs in an additive-additive SNP-interaction model.
| Genotypes | Coding for SNP A | Coding for SNP B | Coding for SNP pair |
|---|---|---|---|
| AA/BB | 0 | 0 | 0 |
| AA/Bb | 0 | 1 | 0 |
| AA/bb | 0 | 2 | 0 |
| Aa/BB | 1 | 0 | 0 |
| Aa/Bb | 1 | 1 | 1 |
| Aa/bb | 1 | 2 | 2 |
| aa/BB | 2 | 0 | 0 |
| aa/Bb | 2 | 1 | 2 |
| aa/bb | 2 | 2 | 4 |
After the SNP pairs were recoded, genomic prediction was performed with all SNP pairs that were selected from each training data set. Therefore, a prediction model was derived using random forest with each training data set (as described above with the ranger function with default settings), the phenotypic values of the corresponding test data set were predicted with the prediction model, and prediction accuracy was estimated as between the predicted and the observed values.
However, since PLINK’s epistasis test is based on linear regression and random forest is a method from machine learning, we developed an alternative for selecting SNP pairs from single SNPs based on machine learning methods. Therefore, we used the information about the variable importance of each single SNP as it was provided by the Boruta function and combined the single SNPs with the highest variable importance to all possible SNP pairs. Subsequently, genomic prediction was performed using all SNP pairs created with the best single SNPs and the resulting prediction accuracy was stored. This process was repeated for the three to 200 single SNPs with the highest variable importance. Finally, the number of single SNPs was determined where the resulting SNP pairs maximised the prediction accuracy. As with the other methods, this method was repeated with each training data set individually to avoid bias and afterwards the median from the ten repetitions was calculated for each number of analysed SNPs. An R script as well as the data from this trial are provided at https://github.com/tmlange/IFS\_SNPpairs.git43 to give researchers the possibility to perform feature selection with SNP pairs based on the variable importance of single SNPs.
The 155 genotypes which carry both known resistances provided raw OD values measured after 90 minutes in the range from 0.1107 to 4 which is the technical limit of the machine. The transformed data were in the range from -7.06 to 12.33. These results show the highest possible variation of virus concentrations that the machine can measure. Thus, although these genotypes were assumed to be resistant at the two known resistance clusters, the resulting data set provides sufficient variance of the resistance levels to perform genomic prediction.
First, genomic prediction was performed using all 9,127 single SNPs that were left after filtering. Performing genomic prediction with the single SNPs with the ten random splits of the data set resulted in a median prediction accuracy of . The median prediction accuracies resulting from genomic prediction using any of the methods described can be seen in Table 2.
| Method | Single SNPs | SNP pairs |
|---|---|---|
| All variables after filtering | 0.146 | 0.191 |
| Subset after feature selection | 0.267 | 0.306 |
Next to genomic prediction with all SNPs that were left after filtering, incremental feature selection was performed to optimise prediction accuracy by selecting a subset of optimal SNPs for genomic prediction. Therefore, Boruta was performed to estimate the variable importance of each SNP. To prevent bias in the data analysis, this analysis was performed in each training data set individually. Subsequently, genomic prediction was performed using random forest.
After performing this analysis, each number of SNPs and the corresponding prediction accuracy was retrieved for each of the ten splits between training and test data set. Figure 1 shows the number of SNPs in the prediction model on the X axis and the corresponding prediction accuracy as the median of the prediction accuracy from the ten repetitions on the Y axis. One can see that the prediction accuracy increases steeply when the first SNPs are included in the prediction model. However, slightly above , the prediction accuracy forms a peak and decreases from there on with the decrease being less steep than the increase at the beginning of the curve.
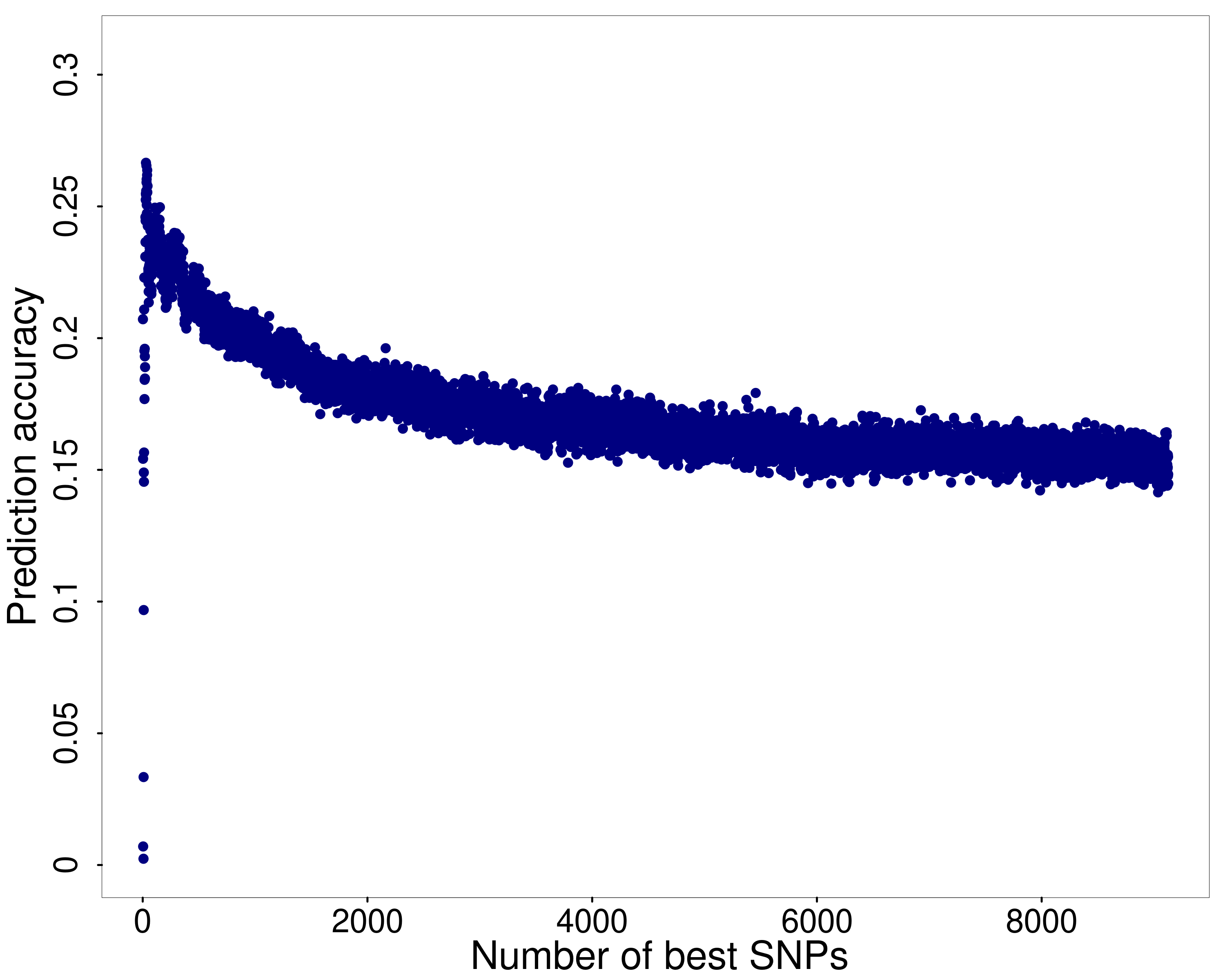
In this way, the optimal set of SNPs for genomic prediction was determined. Thus, the number of SNPs was selected that maximised the median of the prediction accuracy from the ten repetitions. Here, the prediction accuracy maximised if 29 SNPs were included in the prediction model. This resulted in a median prediction accuracy of . However, we found that within the ten training data sets, not all 29 SNPs were the same.
Besides genomic prediction and feature selection with single SNPs, similar approaches have been performed using SNP pairs. To perform genomic prediction with SNP pairs, PLINK’s epistasis test was performed with default settings for each training data set individually. After filtering via PLINK’s epistasis test, only SNP pairs were kept in the data set whose interaction term in a linear regression model led to a value below the default threshold of 0.0001. The resulting sample sizes resulted in 46,556 to 87,529 SNP pairs. Taking the 41 million theoretically possible SNP pairs into consideration, this is a reduction to 0.1% to 0.2%. When genomic prediction was performed with all SNP pairs that were left after filtering using PLINK’s epistasis test with each training set, the median prediction accuracy was .
Besides genomic prediction with all SNP pairs that were left after filtering with PLINK’s epistasis test, feature selection was performed to analyse an optimal subset of SNP pairs for genomic prediction. Therefore, the best single SNPs judged by their variable importance were selected and used to create SNP pairs as described in Table 1. Subsequently, genomic prediction was performed using random forest with the SNP pairs that resulted from the 3, , 200 best single SNPs. Figure 2 displays the median prediction accuracy from the ten repetitions on the Y axis and the corresponding number of single SNPs that make up the SNP pairs on the X axis.
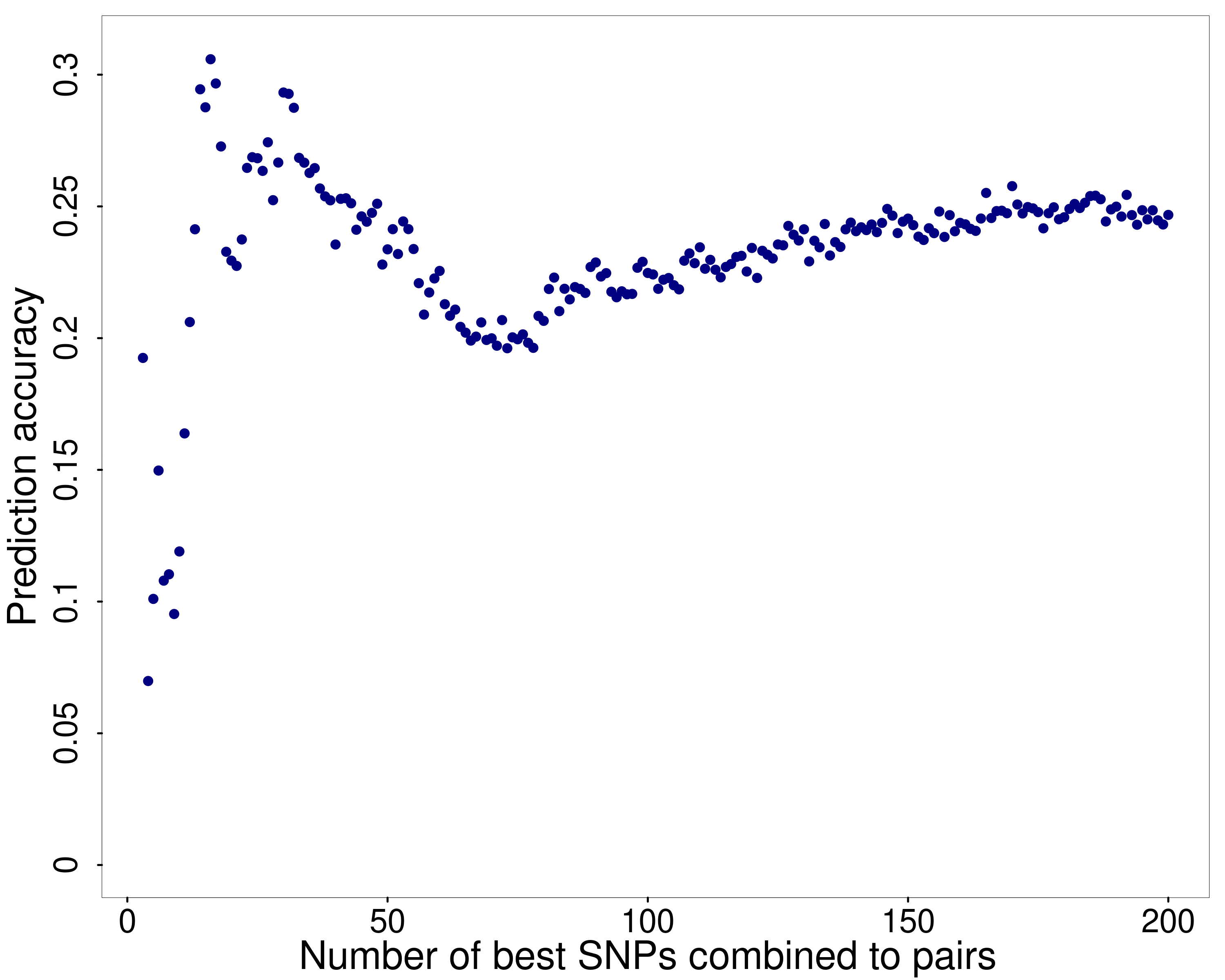
One can see that the prediction accuracy increases steeply when a small number of SNP pairs are included in the prediction model. Similar to Figure 1 that displays the prediction accuracy with the single SNPs, the prediction accuracy with the SNP pairs also forms a peak and decreases from there on. One can see that the peak height is slightly above when SNP pairs are created using the 16 best SNPs.
Considering Figure 2, one can assume that the number of SNP pairs has an effect on the resulting prediction accuracy. Thus, one could assume that the prediction accuracy could be affected by the number of SNP pairs that is selected via PLINK’s epistasis test. Therefore, we have analysed how many SNP pairs were selected via PLINK if the significance threshold was modified. Naturally, the number of selected SNP pairs was reduced if the threshold was decreased and conversely, more SNP pairs were left after filtering if the threshold was increased. However, if subsequently all selected SNP pairs were used for genomic prediction, the resulting prediction accuracy did not seem to be affected (tested for thresholds , , , data not shown). Thus, we conclude that although the number of selected SNP pairs can be easily adjusted in PLINK’s epistasis test, this selection did not affect the resulting prediction accuracy for this data set.
Although rhizomania resistance was assumed to be a quantitative trait caused by major and minor resistance genes,18 to date no attempt of genomic prediction of rhizomania resistance has been performed. We provide a first attempt at predicting the resistance of sugar beet genotypes against rhizomania. To do so, we used a sugar beet population where each genotpye can be assumed to be resistant at Rz1. Furthermore, 155 of the 156 genotypes can be assumed to be resistant at Rz2. Thus, all SNPs in high linkage disequilibrium with either Rz1 or Rz2 should have been removed during SNP pruning. However, the results show that genomic prediction of rhizomania resistance was still possible. In this way, we provide evidence that the genomic architecture of rhizomania resistance is most probably caused by more than two resistance clusters.
To perform genomic prediction, we split the data set ten times randomly into subsets of 80% training data and 20% test data. Performing genomic prediction with all available SNP markers led to a median prediction accuracy of . Moreover, we performed feature selection using methods from machine learning to reduce the number of SNPs in the prediction model to an optimal subset. This method led to a median prediction accuracy of when only the 29 best SNPs were included in the prediction model.
Previous studies of variable importance in genomic prediction have concluded that although SNP interactions can be detected in random forest algorithms, such interactions can be masked by other variables when working with high-dimensional data.62,63 Thus, it could be assumed that SNP interactions are masked when genomic prediction was performed with all single SNPs. Consequently, when the number of SNPs was reduced, SNP interactions were not masked and could be included in the prediction model more efficiently.
Besides genomic prediction using single SNPs, we also present results from genomic prediction using SNP pairs. For this, single SNPs were combined to pairs and the theoretical number of resulting SNP pairs was reduced using PLINK’s epistasis test. Performing genomic prediction with all SNP pairs that were left after filtering resulted in a median prediction accuracy of .
Finally, we used the information about variable importance of each SNP to perform feature selection also with the SNP pairs. Therefore, SNP pairs were created using only the 3, , 200 single SNPs with the highest variable importance and the corresponding prediction accuracy was estimated. This new approach allowed us to perform incremental feature selection using machine learning with SNP pairs. Similar to feature selection with single SNPs, the prediction accuracy for the SNP pairs was maximised if only a certain number of SNP pairs was included in the prediction model. Here, the prediction accuracy was maximised if the prediction model contained SNP pairs from the 16 best single SNPs. This method led to a median prediction accuracy of which was the highest prediction accuracy from all four methods under investigation. These results might indicate that rhizomania is affected by interactions between SNP pairs which are also masked if all SNP pairs are included in the prediction model. Consequently, it might be assumed that not only epistatic effects caused by two genes but also by multiple genes could play a role in rhizomania resistance.
Feature selection was used in recent studies to increase the prediction accuracy of genomic prediction models in man59 and crops.51,54 However, these studies led to heterogeneous results such that no general recommendation can be given regarding feature selection. Here, we show that in case of rhizomania resistance, prediction accuracy could be increased using feature selection. We postulate that the success of implementing feature selection to improve prediction accuracy might be related to epistatic effects that are masked if a large number of SNPs are included in a prediction model. However, we assume the present study alone is not sufficient to underpin this hypothesis. Thus, further research is necessary to study the role of SNP interactions in rhizomania resistance as well as the possibility to improve genomic prediction via feature selection.
Besides improving prediction accuracy of genomic prediction models, other studies used results from feature selection via genomic prediction to determine the association between certain SNPs and the phenotype.54,64 Following this approach, it could be interesting to use the method presented here to select the 29 single SNPs or the 16 SNP pairs, respectively, as being associated with rhizomania resistance. However, we found that the estimation of variable importance did not lead to the same results in each training data set. Consequently, the SNP pairs differed for each training data set as well. Thus, we conclude that the present method can be useful to increase the prediction accuracy of prediction models but is not useful to select certain SNPs or SNP pairs as being associated with the phenotype.
Here, we present a first attempt at genomic prediction of rhizomania resistance in sugar beet. Therefore, we used a sugar beet population with 156 genotypes of which all genotypes can be assumed to be resistant at Rz1 and 155 genotypes can be assumed to be resistant at Rz2. The 155 genotypes that were resistant at Rz2, provided the highest possible variation of virus concentrations that the machine can measure. Moreover, although SNPs in high linkage disequilibrium with Rz1 and Rz2 were removed during SNP pruning, genomic prediction was possible with the genomic data. If rhizomania resistance was caused only by the two known resistance clusters, this should not be the case.
To perform genomic prediction, we have used single SNPs as well as SNP pairs. In the provided data set, the genomic prediction using SNP pairs led to higher prediction accuracy than the genomic prediction using single SNPs. These results lead to the conclusion that epistatic effects might affect rhizomania resistance and that the usage of SNP pairs can include these effects more efficiently in the prediction model.
Moreover, we have shown that a selection of the “best” SNPs increased prediction accuracy even further. It was concluded in former studies that although random forest can detect SNP interactions, such interactions can be masked by other variables in high-dimensional data.62,63 In this way, our results fit to the conclusions from these studies since the prediction accuracy was increased if only a subset of all available SNPs was used for genomic prediction. Moreover, our results indicate that the variable importance that was estimated using Boruta, hence, random forest, also included information about SNP interactions. Thus, we conclude that a reduction of the SNP number via the variable importance enables the random forest algorithm to incorporate SNP interactions better into the prediction model.
Furthermore, we have also performed feature selection with the SNP pairs to reduce the data set to a certain subset of optimal SNP pairs. The inclusion of a subset of SNP pairs in the prediction model increased the prediction accuracy compared to the prediction model that included all SNP pairs that were left after filtering via PLINK’s epistasis test. Following the conclusion with the single SNPs, this might indicate that rhizomania resistance is caused by interactions of more than two genes and that the interaction of SNP pairs might be similarly masked if a large number of SNP pairs is included in the prediction model.
All in all, the optimisation of the prediction model increased the median prediction accuracy with the ten repetitions that are provided here. These results make us assume that rhizomania resistance could be caused by a multitude of genes which interact and that the implementation of such interactions in a prediction model can increase prediction accuracy. However, further research in this regard is necessary. To encourage researchers to perform feature selection with SNP pairs in their own studies, we have published an R script as well as the data from this trial at https://github.com/tmlange/IFS_SNPpairs.git.43
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)