Keywords
Spinal meningioma; pediatric meningioma; back pain; gait disturbance
This article is included in the Oncology gateway.
Spinal meningioma; pediatric meningioma; back pain; gait disturbance
In this revised version, we have expanded the case presentation to include a detailed timeline of symptom progression, with objective measures such as the Modified Ashworth Scale for spasticity and additional findings on posterior column involvement. MRI description has been refined to clarify the extramedullary location, absence of intramedullary involvement, and to address the differential diagnosis more accurately. Surgical details have been updated to discuss the extent of laminectomy, with commentary on alternative approaches and potential implications for postoperative spinal stability in pediatric patients.
The discussion has been fully reorganized into thematic sections focusing on pathogenesis and epidemiology, clinical presentation and differential diagnosis, imaging and diagnostic features, surgical management and outcomes, and long-term follow-up considerations. References have been revised for accuracy, with removal of irrelevant surgical approaches and correction of statements not supported by cited literature.
Follow-up details now provide a more objective neurological assessment; however, the patient was lost to follow-up before postoperative MRI could be obtained. Figures have been updated to include representative MRI sequences that illustrate the lesion characteristics described in the text. Minor language edits were also made to improve clarity, precision, and conciseness throughout the manuscript.
See the authors' detailed response to the review by Kautilya R Patel
1. Spinal pediatric meningiomas are rare and can mimic various conditions, making diagnosis particularly challenging.
2. MRI plays a crucial role in differentiating between potential diagnoses. However, histopathological confirmation is essential for diagnosing meningiomas and guiding appropriate management.
3. Detailed history, thorough physical examination, and genetic screening are crucial to rule out associations with genetic conditions, particularly NF2, which necessitates a different management approach and genetic counseling.
4. Surgery aiming for total tumor resection is the first-line treatment for spinal pediatric meningiomas. Unfortunately, the post-operative course in this population is often complicated by higher recurrence rates and additional complications.
Gait abnormalities are common in children and can result from various conditions, including cerebral palsy, neuromuscular disorders, and spinal cord tumors, such as spinal meningiomas. Spinal meningiomas account for less than 10% of central nervous system tumors in the pediatric population.1
Pediatric meningiomas (PMs) are an even rarer entity, accounting for less than 5% of all meningiomas, and spinal PMs are estimated to be exceedingly rare.2
Spinal meningiomas are characterized by their intradural, extramedullary growth,1 often resulting in a mass effect on the spinal cord that can lead to significant spinal cord compression and related complications.3
Surgical resection remains the cornerstone of treatment and is generally safe when performed in specialized centers. However, management in pediatric patients requires particular attention given the rarity of the condition, its often atypical presentation, and the potential association with genetic disorders such as neurofibromatosis type 2 (NF2).2
Although surgical techniques mirror adult approaches, pediatric meningioma resection is uniquely challenging due to smaller anatomical dimensions, higher recurrence rates, and greater complication risk.4 Radiotherapy is typically reserved for selected cases, especially in recurrent or NF2-related tumors, due to the long-term risks associated with irradiation of the developing nervous system.2
We report a case of pediatric spinal meningioma revealed by gait disturbance. We emphasize clinical and imaging features, as well as therapeutic management of spinal meningiomas.
A 15-year-old male presented with a one-year history of progressively worsening thoracic back pain. The pain had an insidious onset, was dull and persistent, and remained localized to the thoracic spine, without any radiation. Initially, the pain was mild but gradually intensified over time, affecting daily activities. About one month before presentation, the patient noticed progressively worsening gait instability, marked by unsteadiness and repeated balance loss, without dizziness or vertigo. In the week leading up to hospitalization, he developed significant lower extremity weakness, causing ambulatory difficulty beyond 100 meters due to rapid fatigue and a sensation of heaviness in the legs. No sensory complaints were reported. He denied any history of trauma, constitutional symptoms (such as fever or weight loss), or neurological symptoms like bowel or bladder dysfunction, seizures, or headaches. Family history was negative for neurofibromatosis, malignancy, Gorlin syndrome, or familial meningiomatosis.
Preoperative physical examination revealed a spastic gait with increased lower limb muscle tone (Modified Ashworth Scale (MAS) 3/4)5 but preserved strength (5/5) in all limbs, and no sensory deficits. Deep tendon reflexes were brisk and symmetrical in the lower extremities, and the Babinski sign was bilaterally positive, consistent with upper motor neuron involvement. Superficial reflexes were diminished, with bilateral abolition of abdominal cutaneous reflexes. Gait examination revealed marked unsteadiness with balance impairment, attributable to spasticity rather than vestibular or cerebellar dysfunction. He had no posterior column involvement with normal vibration perception, unimpaired light touch discrimination, and preserved proprioception, including hallux position sense. There was no spinal tenderness on palpation or percussion, and no signs of meningeal irritation, spinal stiffness, or joint involvement. Cardiovascular and pulmonary examinations were unremarkable. Additionally, no cutaneous lesions suggestive of neurocutaneous syndromes were observed.
C-reactive protein, calcium level, liver tests, and renal function were within normal ranges. The complete blood count was without abnormalities.
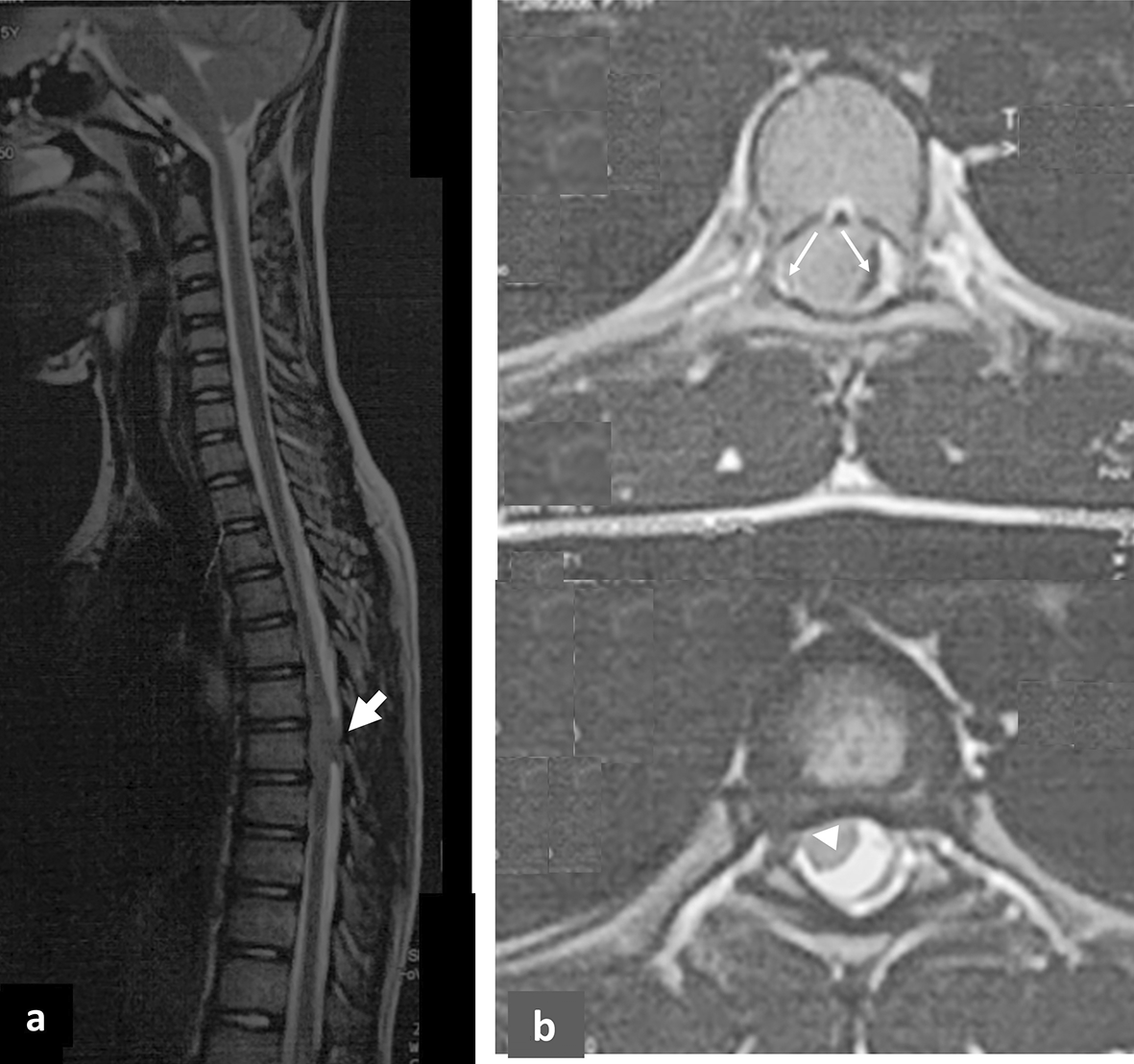
Given the presence of neurological signs, a spine MRI was performed showing a posterior intradural, left-sided extramedullary lesion at the T6 and T7 vertebral levels, characterized by an extramedullary, well-circumscribed, homogeneous, and lobulated appearance. The lesion measured approximately 1.8 × 1.2 × 0.9 cm and exhibited iso-intensity on T1-weighted images and hyperintensity on T2-weighted images, with homogeneous and marked enhancement after contrast administration. The mass exerted a dorsal mass effect, resulting in spinal cord compression [ Figure 1a and 1b]. These imaging findings were initially suggestive of meningioma.
The patient underwent posterior spinal surgery, including a wide laminectomy spanning T5, T6, and T7 [Figure 2].
Upon opening the dura mater, a whitish, firm mass compressing the adjacent spinal cord was observed. The tumor displayed a well-defined cleavage plane with the dura mater, with only minor adhesions to the adjacent arachnoid. Complete resection of the tumor was achieved.

Histopathological examination revealed a proliferation of meningothelial cells with poorly defined cytoplasmic borders, creating a syncytial appearance and exhibiting cytological pleomorphism. Numerous psammoma bodies were observed throughout the specimen, with no evidence of anaplasia or tumor necrosis [ Figure 3]. These findings were consistent with a WHO Grade 1 psammomatous meningioma.

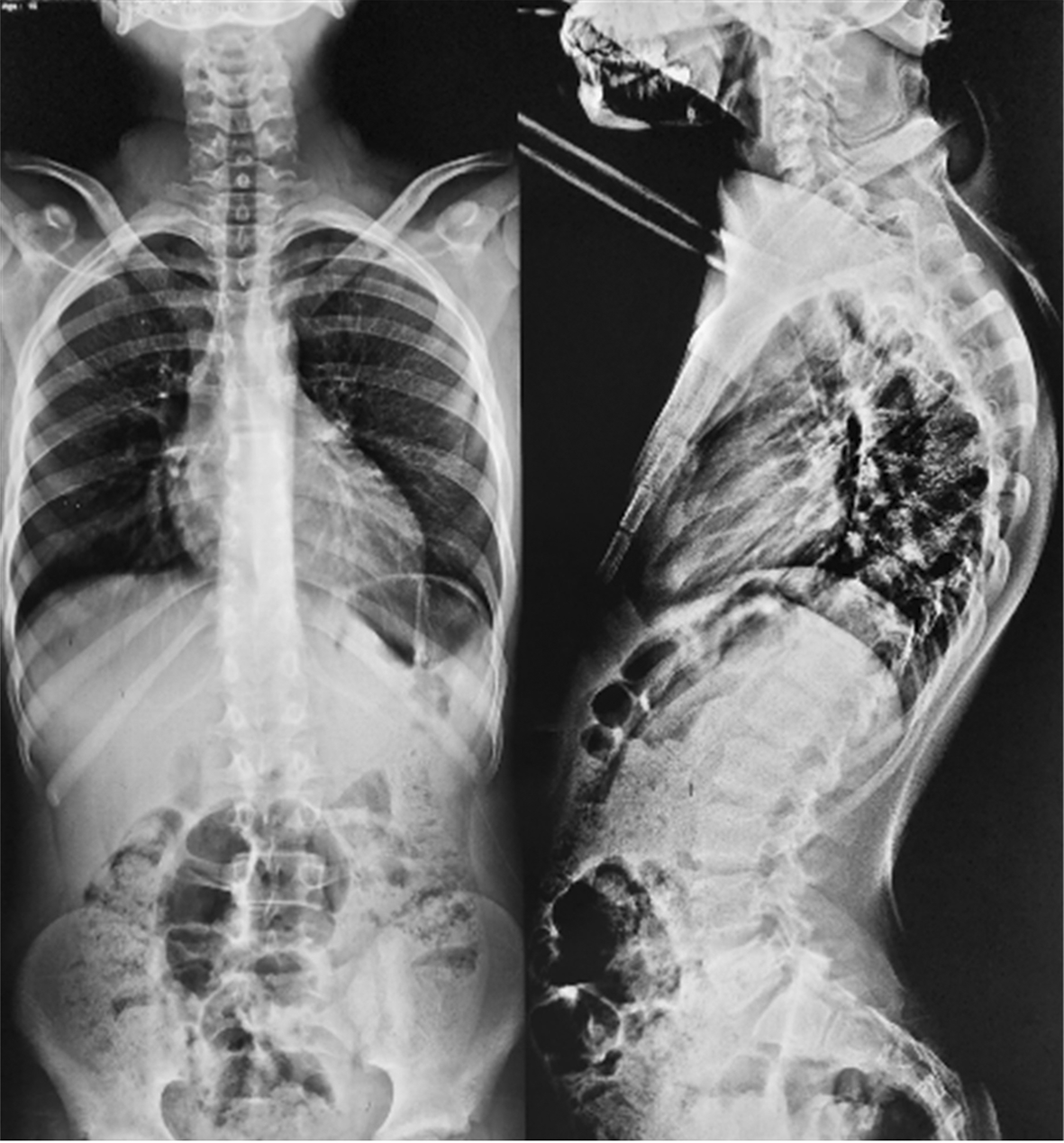
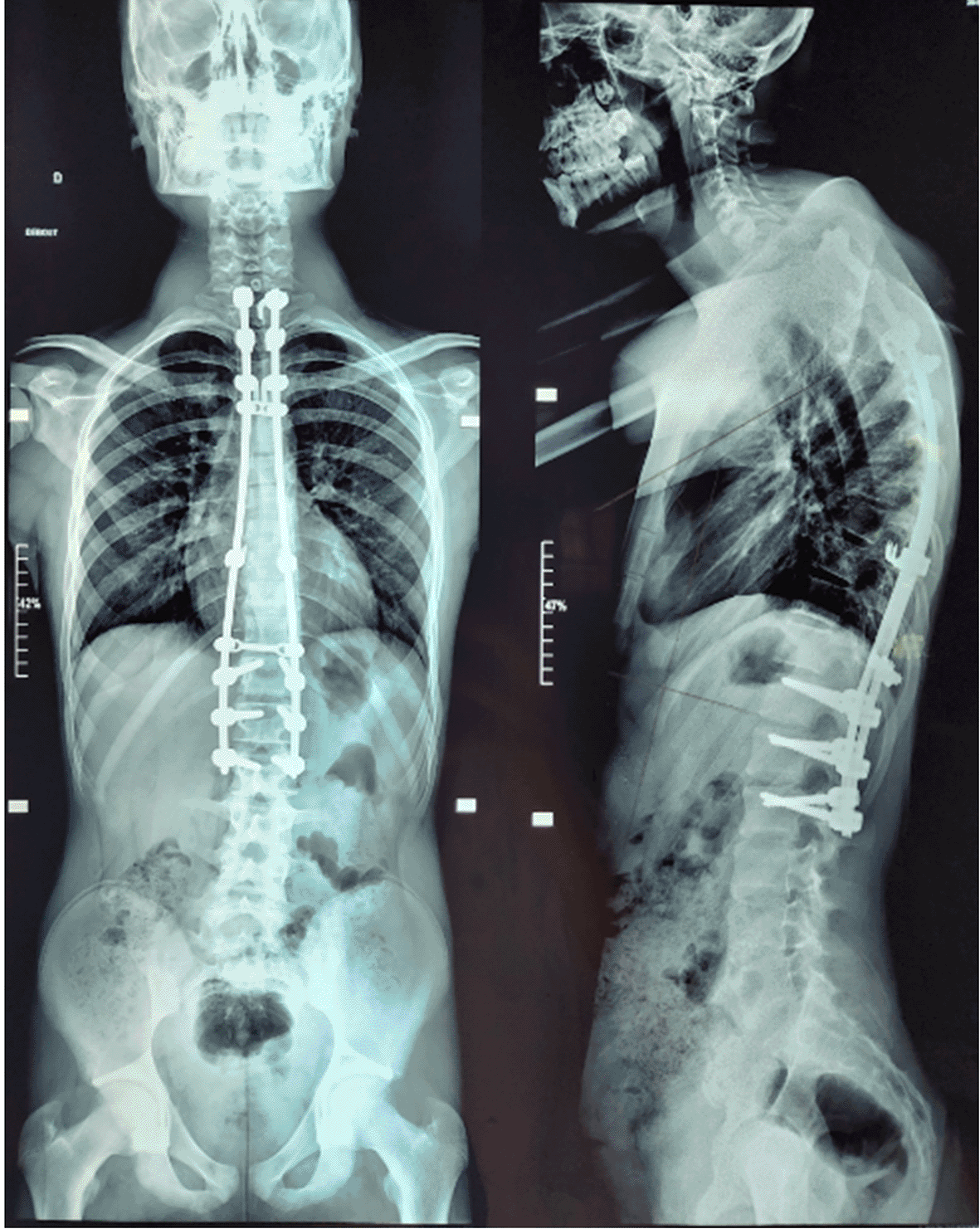
The postoperative course was complicated two months after surgery by severe hyperalgesic back pain and progressive dorsal hyperkyphosis [Figure 4]. Attributed to the extensive laminectomy, ultimately requiring posterior thoracic arthrodesis [Figure 5]. At latest follow-up, neurological examination showed preserved motor strength (5/5 in lower limbs), moderate spasticity (MAS 2/4), brisk symmetrical reflexes with bilateral Babinski signs, intact superficial sensation, and no autonomic dysfunction, though mid-thoracic tenderness persisted without instability.
At two-year postoperative follow-up, the patient continues to demonstrate sustained functional gains through neurorehabilitation, with significant reduction in pain levels and maintained improvement in postural alignment.
We present the case of a pediatric spinal meningioma revealed by an insidious onset of gait disturbance, thoracic back pain, and lower limb weakness. Meningiomas are typically slow-growing tumors originating from the meninges.3
Similarly to intracranial meningiomas, spinal meningiomas arise from arachnoid cap cells. Abnormal proliferation of these cells leads to the formation of intradural extramedullary masses. Spinal meningiomas represent 5 to 10% of all meningiomas and 25 to 45% of all intraspinal tumors.1,2 Spinal meningioma typically occurs in the thoracic spine since arachnoid cap cells are predominantly located at this spinal level.3,4
Pediatric meningiomas are scarce, accounting for less than 5% of pediatric spinal tumors.2 Unlike in adults, pediatric meningiomas seem to be less influenced by hormonal factors, which may explain the similar gender distribution observed in this population.2,3,5
Meningiomas are graded by the World Health Organization (WHO) into three grades based on their histological features, such as mitotic rate, cellular atypia, and brain invasion. Grade I meningiomas are benign characterized by a low mitotic rate (<4 per 10 high-power fields) and the absence of brain invasion, and include a psammomatous subtype,6 as seen in our patient. The same grading system applies to PMs.
In contrast to adults, an association between pediatric meningiomas and type 2 neurofibromatosis (NF2) has been reported in 13% of cases and even in more than 50% of patients with intracranial meningiomas.7 The NF2 gene mutation is responsible for abnormal cell proliferation and tumor formation. NF2-related meningiomas are characterized by an earlier onset, more rapid growth, atypical histological features, and higher WHO grades.2,3,7 Pediatric meningiomas may also be associated with other genetic syndromes, such as Gorlin syndrome (basal cell nevus syndrome) and familial meningiomatosis. However, our patient had no personal or family history of these conditions. In a study of CT scans from 82 patients with Gorlin syndrome, 5% showed radiological features suggestive of meningioma.8
Spinal meningiomas are characterized by their intradural, extramedullary growth, typically affecting the thoracic spine,3,6 as seen in our patient. While asymptomatic spinal meningiomas usually follow a slow, linear growth pattern, symptomatic tumors exhibit a rapid exponential pattern,3,5 subsequently leading to varying degrees of spinal cord compression ranging from insidious motor dysfunction and sensory dysfunction to gait disturbances and difficulty walking, as observed in our patient. Symptoms generally worsen progressively over time as the tumor continues to expand, exerting increased pressure on the spinal cord. Our patient’s insidious onset of thoracic back pain, followed by gait instability and spastic paraparesis, typifies the progressive compression seen in symptomatic tumors.
In advanced stages, severe spinal cord compression can disrupt autonomic functions, resulting in bowel and bladder disturbances, which are reported in approximately 36% of patients, and irreversible neurological damage.2,5 Chronic cases often present with persistent back pain, as was evident in our patient, which may be associated with radiculopathy or evolve into a more debilitating condition. Longstanding tumor presence can also contribute to spinal deformities, such as kyphosis.5
MRI remains the gold standard for diagnosis and surgical planning. Pediatric spinal meningiomas typically appear as well-circumscribed, intradural extramedullary masses, iso- to hypointense on T1, hyperintense on T2, and exhibit homogenous post-contrast enhancement with a dural tail sign.3,4 Unlike intramedullary lesions, meningiomas do not expand the cord and are eccentrically located.
The patient’s presentation initially suggested transverse myelitis. Nevertheless, this diagnosis was less likely given the subacute symptom progression and absence of sensory and autonomic dysfunction. The transverse myelitis, which is an intramedullary inflammatory process, was definitively excluded by MRI findings.7
While spinal meningiomas classically present with distinctive MRI features, pediatric cases present unique diagnostic challenges. Indeed, these tumors are scarce in children, which often lowers clinical suspicion. Furthermore, approximately 30% exhibit atypical radiological features1–4 such as cystic changes or heterogeneous enhancement that mimic more common pediatric lesions like ependymomas or nerve sheath tumors. Although our preoperative imaging strongly suggested meningioma, histopathological confirmation remained imperative.
Neoplastic etiologies are among the most frequent causes of spinal cord compression in the pediatric population, commonly involving ependymomas, astrocytomas, gliomas, and meningiomas. MRI findings are typical for meningioma and distinguish it from intramedullary tumors such as gliomas, astrocytomas, and ependymomas, which usually present with ill-defined margins, cord expansion, and absence of a dural tail. Gliomas and astrocytomas often involve multiple spinal segments and demonstrate diffuse intramedullary signal changes. Ependymomas, while sometimes well-circumscribed, are also intramedullary and may feature a “cap sign” on T2-weighted images or an associated syrinx, neither of which was observed in our case.8–10
Tethered cord syndrome can mimic spinal meningioma due to gait abnormalities and progressive motor dysfunction of the lower limbs. However, no foot deformities or cutaneous markers such as sacral dimples, lipomas, or hairy patches along the midline of the back were observed on examination. MRI ruled out this diagnosis, as it did not reveal a low-lying conus medullaris, thickened filum terminal, or evidence of spinal cord tension.11
Meningiomas are classified by the World Health Organization (WHO) into three grades based on their histological features, such as mitotic rate, cellular atypia, and brain invasion. Grade I meningiomas are benign, characterized by a low mitotic rate (<4 per 10 high-power fields) and the absence of brain invasion, and include a psammomatous subtype,6 as seen in our patient.
Management of meningioma is largely dependent on its symptomatic status.3 Asymptomatic meningiomas can often be managed with routine observation and imaging.3,6 Conversely, symptomatic and rapidly enlarging meningiomas, typically require surgical resection as the primary approach.3,6 Gross total resection is the treatment of choice for symptomatic spinal meningiomas and can generally be performed safely, even in pediatric patients.3,4 Intraoperatively, our patient’s tumor displayed a clear cleavage plane, permitting complete excision.
Unfortunately, the postoperative course in pediatric patients is marked by a higher incidence of spinal instability, especially following extensive laminectomies,2,6,12 necessitating spinal fusion or stabilization procedures, as was required in our patient.
While our initial approach utilized a wide laminectomy for optimal tumor access, alternative techniques, such as a single-level laminectomy with undermining of adjacent levels, hemilaminectomy, or laminoplasty, may offer superior spinal stability preservation in pediatric patients, potentially reducing postoperative kyphosis risk.12–14 This consideration is particularly relevant for young patients with developing spines, where extensive bone removal can predispose to deformity.
Adjuvant radiotherapy (RT) may be recommended in cases of recurrent tumors, non-resectable, growing lesions, or when gross total resection is not achievable.3,6 However, RT is generally avoided in pediatric patients due to the heightened risk of secondary malignancies and long-term neurocognitive sequelae.2,6,8
To date, novel pharmacotherapies, including multikinase inhibitors targeting vascular endothelial growth factor receptors and bevacizumab, have shown promising results; however, consensus guidelines have yet to be established.15 Interestingly, radionuclide therapies targeting somatostatin receptors are under exploration for the management of refractory meningiomas, demonstrating great potential.12
Recurrence is rare following gross total resection of WHO Grade I meningiomas, but long-term surveillance is essential, especially in children. Recommended follow-up includes MRI every 6 months for the first 5 years, followed by annual imaging.5,6,12
The diagnosis of spinal tumors should be considered in children with gait disturbances.
Pediatric meningiomas present with a spectrum of symptoms, ranging from back pain to severe spinal cord compression and even spinal deformities. MRI typically shows a well-defined and circumscribed extramedullary appearance with the characteristic dural tail sign on post-contrast images. A total resection is considered the gold standard. Radiotherapy is reserved for specific scenarios, including recurrence, subtotal resection, inoperability, or high-grade tumors.
Consent: Written informed consent for publication of their clinical details was obtained from the patient’s legal guardians. All figures pertain to our patient and have not been previously published. We have obtained formal written consent from the patients’ legal guardians for their publication.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)