Sandiford SL, Noble SAA, Pierre SA et al. The first mitochondrial genome of Haemagogus equinus from Jamaica [version 2; peer review: 2 approved, 1 approved with reservations]. F1000Research 2025, 13:1504 (https://doi.org/10.12688/f1000research.159115.2)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
1Department of Basic Medical Sciences, Pharmacology and Pharmacy Section, Faculty of Medical Sciences, The University of the West Indies Mona, Kingston, Jamaica 2Mosquito Control and Research Unit, The University of the West Indies, Mona, Kingston, Jamaica 3Department of Microbiology, Faculty of Medical Sciences, The University of the West Indies Mona, Kingston, Jamaica 4Department of Life Sciences, Faculty of Science & Technology, The University of the West Indies Mona, Kingston, Jamaica 5The W. Harry Feinstone Department of Molecular Microbiology and Immunology, Johns Hopkins Malaria Research Institute, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland, USA
Simone L. Sandiford
Roles:
Conceptualization,
Project Administration,
Resources,
Supervision,
Writing – Original Draft Preparation,
Writing – Review & Editing
Simmoy A. A. Noble
Roles:
Investigation,
Resources,
Writing – Review & Editing
Samra A. Pierre
Roles:
Investigation,
Resources,
Writing – Review & Editing
Douglas E. Norris
Roles:
Funding Acquisition,
Resources,
Writing – Review & Editing
Renee Ali
Roles:
Data Curation,
Formal Analysis,
Methodology,
Resources,
Visualization,
Writing – Original Draft Preparation
In the Americas, the expansion in incidence of arboviral infections including Mayaro virus (MAYV) has drawn attention to the resurgence of viruses associated with understudied arthropods. Mosquitoes belonging to the genus Haemagogus are generally geographically restricted to the forests of Central and South America and the Caribbean and are the known sylvan vectors for yellow fever virus and emerging MAYV. With an established population in Jamaica, Haemagogus equinus has been reported to be well-adapted to oviposition in artificial containers close to human populations. Its role in arboviral transmission however is not fully understood. Given the dearth of genetic information and the difficulty in morphologically identifying cryptic features in species belonging to this genus, we report the first mitochondrial genome of Hg. equinus. Using a genome skimming approach, two Hg. equinus mosquito specimens were sequenced using the Illumina Novaseq 6000 platform. A representative mitogenome of 16,471 bp, 80.7% AT and 37 genes was assembled using NOVOplasty. Phylogenetic analysis placed Hg. equinus in the Albomaculatus section of the Haemagogus subgenus supporting previously described taxonomic studies.
Corresponding author:
Simone L. Sandiford
Competing interests:
No competing interests were disclosed.
Grant information:
SAAN was supported in part by the University at Buffalo Clinical and Translational Science Institute award UL1TR001412 and the Global Infectious Diseases Research Training Program award D43TW010919. DEN and RA were supported in part by funding from the NSF-Accelerator Project D-688: Computing the Biome (2134862), Johns Hopkins Malaria Research Institute and Bloomberg Philanthropies.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The revised version of this article now includes additional details regarding the molecular analysis which was conducted. Furthermore, a higher resolution version of figure 1 has been provided and figure 2 has been revised with the addition of Ae. aegypti as an outgroup in the phylogenetic tree, and the Haemagogus and Conospotegus subgenera are now both clearly displayed in the figure. Minor revisions have also been made to the figure 2 legend to provide further clarity regarding the use of Psorophora and Aedes as outgroups for the phylogenetic tree. Information pertaining to the similarity between the two sequenced specimens has also been added to the results section.
The revised version of this article now includes additional details regarding the molecular analysis which was conducted. Furthermore, a higher resolution version of figure 1 has been provided and figure 2 has been revised with the addition of Ae. aegypti as an outgroup in the phylogenetic tree, and the Haemagogus and Conospotegus subgenera are now both clearly displayed in the figure. Minor revisions have also been made to the figure 2 legend to provide further clarity regarding the use of Psorophora and Aedes as outgroups for the phylogenetic tree. Information pertaining to the similarity between the two sequenced specimens has also been added to the results section.
Mosquitoes of the genus Haemagogus (Hg.) Williston, 1896 are endemic to tropical rainforests, open deciduous forests and mangroves in Latin America and the Caribbean.1 The genus is divided into two subgenera (Haemagogus and Conopostegus), with the Haemagogus subgenus split into three sections (Albomaculatus, Splendens, Tropicalis).1 Being the primary sylvatic vectors for yellow fever virus (YFV)1 and the emerging arboviral threat Mayaro virus (MAYV),2 species in this genus of mosquitoes are of significant medical importance in the region. Despite this, many species remain understudied and their role in disease transmission has not been clearly defined. In Jamaica, Haemagogus equinus Theobald, 1903 is presently the only known species on the island. Established populations of Hg. equinus have also been identified in southern Texas in the United States of America, Mexico, Belize, Guatemala, El Salvador, Honduras, Nicaragua, Costa Rica, Panama, Colombia, Guyana, Venezuela and Trinidad and Tobago.1
While the vectorial capacity of Hg. equinus has not been fully elucidated, laboratory and transovarial transmission of YFV3–6 along with natural infections among wild mosquitoes have been reported.7,8 In spite of this, its importance in arboviral transmission in some localities remains uncertain.9 Although Hg. equinus primarily oviposits and larvae develop in tree holes and bamboo internodes,1 the species is very adaptable and can also utilize rock holes,10 domestic containers and used tires.11,12 With multiple reports of MAYV in neighbouring Haiti,13–15 there is need for a greater understanding of the role of this sylvatic vector in arbovirus transmission. To facilitate this, accurate identification of all life stages of field collected specimens is crucial. However, impediments such as a lack of taxonomic expertise coupled with poor quality samples and a paucity of reference molecular data continue to hamper these efforts in Jamaica.
Few studies have employed genomic sequencing to investigate the molecular taxonomy and phylogenetic relationships of Hg. equinus mosquitoes. Currently, only partial sequences of cytochrome oxidase I and other mitochondrial or nuclear genes are available from NCBI’s GenBank or BOLD systems databases.16–21 The complete mitochondrial genome (mitogenome) is increasingly being used for evolutionary and phylogeny studies due to its maternal inheritance, simple genomic organization, relative abundance in tissues and absence of recombination.22 In most metazoans the mitogenome is highly conserved and comprises of 13 protein coding genes, 22 transfer RNAs and two ribosomal RNAs in addition to a large single non-coding region important for replication and transcription.23 At present, mitogenomes of only five species from the genus Haemagogus are available.24,25 Considering the significance of mitogenomes when conducting phylogenetic and taxonomic studies, we describe for the first time the characterization of the mitogenome of Hg. equinus using a genome skimming approach and the phylogenetic comparison with other Haemagogus
taxa.
Methods
The Hg. equinus samples (n = 2) sequenced were collected in Mona, Jamaica (18.0061364°N, -76.7515125°W) in May and December 2023 in an area characterized by the predominant growth of banana plants and adjacent to a forested area. Briefly, BG sentinel traps (BioGents, Regensburg, Germany) baited with two pounds of dry ice (without lure) were placed overnight from 1400 to 1000 hr and collected mosquito specimens were sorted and morphologically identified using taxonomic keys.26 Single specimens were stored in tubes containing silica and shipped to the Johns Hopkins Bloomberg School of Public Health for molecular analysis as described in detail in.27 Briefly, each mosquito specimen was homogenized in PK buffer (Applied Biosystems, Waltham, USA) containing proteinase K (Applied Biosystems, Waltham, USA) and incubated at 56°C. DNA was then extracted using the Qiagen DNeasy Blood and Tissue Kit (Hilden, Germany) and quantified using the Qubit dsDNA assay kit (ThermoFisher, Waltham, USA) prior to library construction and Illumina sequencing which was conducted at SeqCenter (Pittsburgh, USA).
The mitogenome was assembled as described for African anophelines using NOVOPlasty (RRID:SCR_017335) version 4.3.127,28 and automatic annotations performed with MITOS on the galaxy platform under the invertebrate genetic code.29,30 Geneious Prime (RRID:SCR_010519) version 2023.2.1 (Biomatters, Auckland, Australia) was utilized for manual adjustments of start and stop codons to match reference Haemagogus mitogenomes in the GenBank repository.
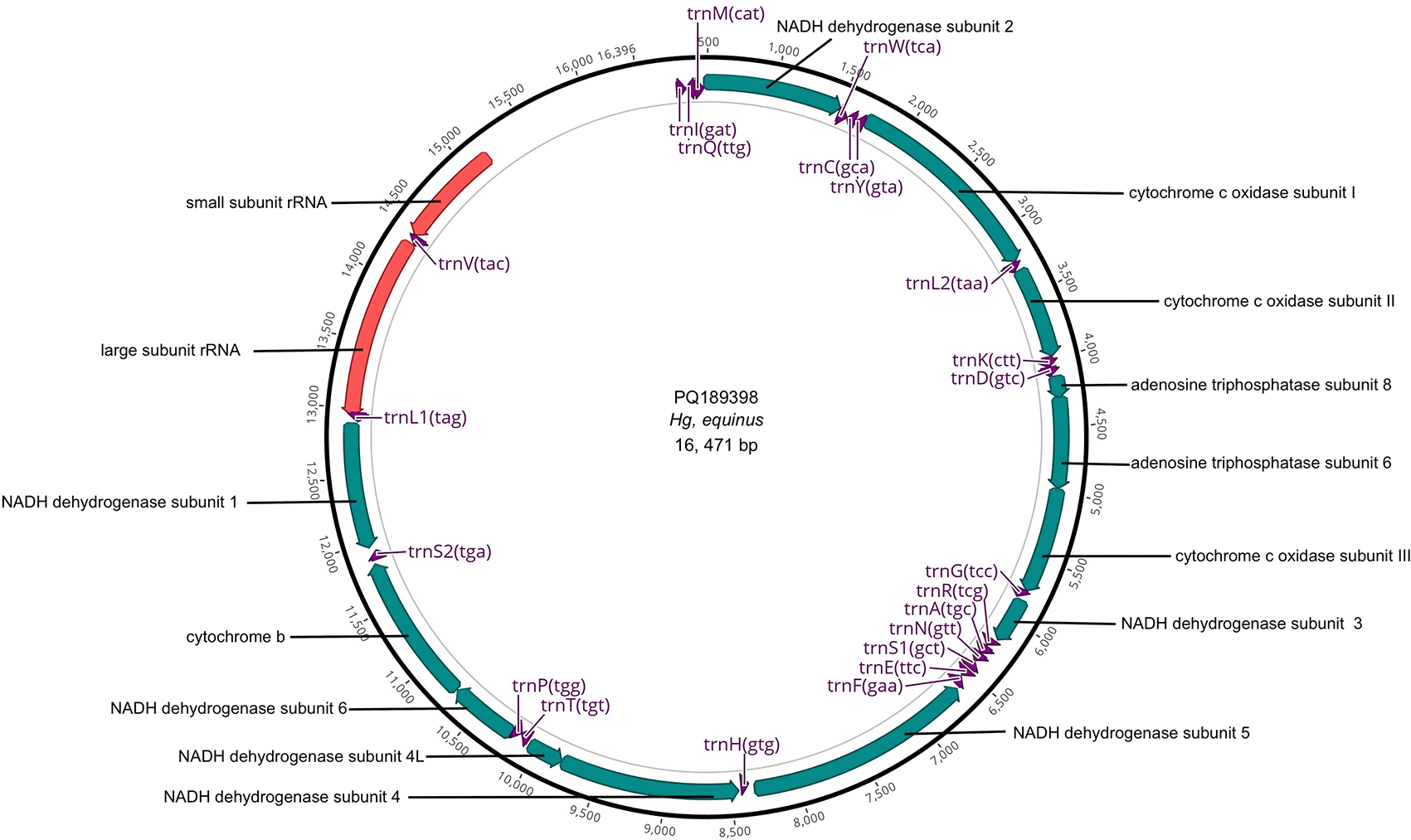
Figure 1 illustrates a representative map of the mitochondrial sequences and annotations submitted to GenBank.
Figure 1. Mitogenome map of Hg. equinus with annotated genes.
Nine mitogenomes (two sequenced from this study and seven available from GenBank) were used for phylogenetic analysis. The 13 protein coding genes of these mitogenomes were extracted with Geneious Prime and aligned by MAFFT version 7.490 into a single matrix. jModelTest version 2.1.10 identified the best fit substitution model and phylogenetic analyses were performed using maximum likelihood in MEGA (RRID_SCR_023017) version 11 with 1000 bootstrap replicates.
Results
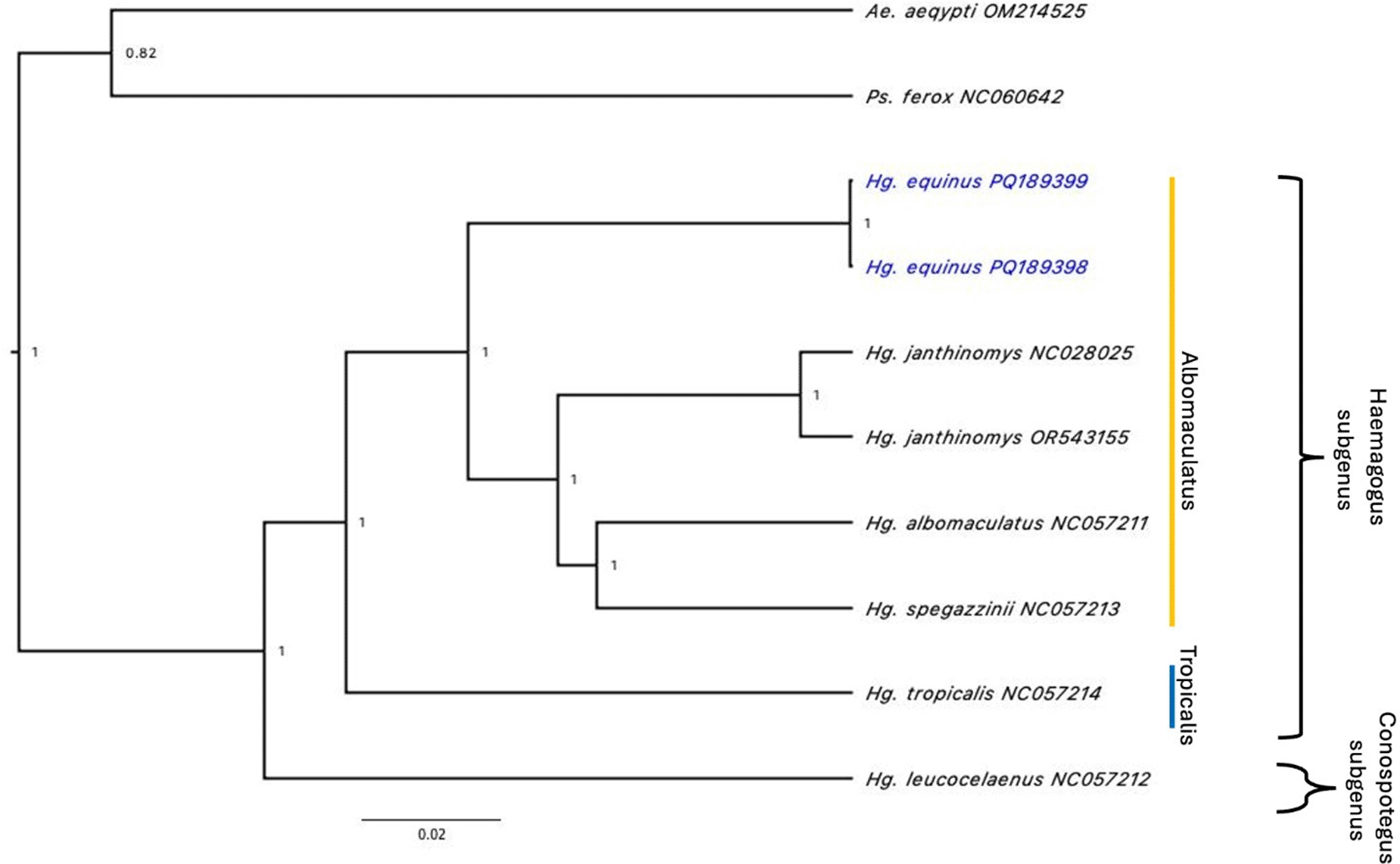
Sequencing of the two Hg. equinus specimens resulted in a mean of 26,728,491 million reads and of these approximately 69,261 reads were utilized for assembling each mitogenome. The two specimens shared 98.84% similarity. Two ribosomal RNAs, 22 transfer RNAs and 13 protein coding genes were detected in the two Hg. equinus mitochondrial genomes (GenBank accession numbers PQ_189398, PQ_189399). The cytochrome c oxidase I (COI) segment covering 1691-3221 bp was 99.59 % comparable to a COI sequence for Haemagogus spegazzinii Brèthes, 1912 (YP_010155459) retrieved from GenBank. Similar to the six Haemagogus mitochondrial genomes available in the GenBank database, the representative mitogenome from our sequencing efforts (PQ_189398) has an A+T proportion of 80.7% and length of 16,471 bp. The Maximum Likelihood phylogenetic tree places it in the Albomaculatus section, separated from other Haemagogus mitogenomes (
Figure 2).
Figure 2. Maximum likelihood tree using the General Time Reversible (GTR + G + 1) substitution model based on the 13 concatanated protein coding genes of Hg. equinus, six Haemagogus species and Psorophora ferox von Humboldt, 1819 and Aedes aegypti Linnaeus, 1762 as outgroups. Haemagogus, Psorophora and Aedes are all members of the tribe Aedini.
These findings provide the basis for the development of more accurate molecular tools which can be used for identification of Hg. equinus mosquitoes. Furthermore, by populating molecular repositories with genomic data from Jamaican mosquitoes, their phylogenetic and evolutionary status will be better understood.
Simmoy A.A. Noble is a Global Infectious Diseases Scholar and received mentored research training in the development of this manuscript.
References
1.
Arnell JH:
Mosquito studies (Diptera, Culicidae). XXXII. A revision of the genus Haemagogus.
Contributions of the American Entomological Institute.
1973; 10: ii + 1–ii + 174.
2.
Pan American Health Organization:
Epidemiological Alert: Mayaro Fever.
(accessed on 15 August).
Reference Source
3.
Waddell MB, Taylor RM:
Studies on Cyclic Passage of Yellow Fever Virus in South American Mammals and Mosquitoes: Marmosets (Callithrix aurita) and Cebus Monkeys (Cebus versutus) in Combination with Aedes aegypti and Haemagogus equinus.
Am. J. Trop. Med.
1945; s1-25: 225–230. Publisher Full Text
4.
Waddell MB, Taylor RM:
Studies on the Cyclic Passage of Yellow Fever Virus in South American Mammals and Mosquitoes: III. Further Observations on Haemagogus equinus as a Vector of the Virus.
Am. J. Trop. Med.
1947; s1-27: 471–476. Publisher Full Text
5.
Waddell MB:
Comparative Efficacy of Certain South American Aëdes and Haemagogus Mosquitoes as Laboratory Vectors of Yellow Fever.
Am. J. Trop. Med.
1949; 29: 567–575. PubMed Abstract
| Publisher Full Text
6.
Dutary BE, Leduc JW:
Transovarial transmission of yellow fever virus by a sylvatic vector, Haemagogus equinus.
Trans. R. Soc. Trop. Med. Hyg.
1981; 75: 128. PubMed Abstract
| Publisher Full Text
7.
De Rodaniche E, Galindo P:
Isolation of yellow fever virus from Haemagogus mesodentatus, H. equinus and Sabethes chloropterus captured in Guatemala in 1956.
Am. J. Trop. Med. Hyg.
1957; 6: 232–237. PubMed Abstract
| Publisher Full Text
8.
De Rodaniche E, Galindo P, Johnson CM:
Isolation of yellow fever virus from Haemagogus lucifer, H. equinus, H. spegazzinii falco, Sabethes chloropterus and Anopheles neivai captured in Panama in the fall of 1956.
Am. J. Trop. Med. Hyg.
1957; 6: 681–685. PubMed Abstract
| Publisher Full Text
9.
Chadee DD, Hingwan JO, Persad RC, et al.:
Seasonal abundance, biting cycle, parity and vector potential of the mosquito Haemagogus equinus in Trinidad.
Med. Vet. Entomol.
1993; 7: 141–146. PubMed Abstract
| Publisher Full Text
10.
Chadee DD:
Rock hole breeding Haemagogus mosquitoes on Monos Island, Trinidad, West Indies.
Mosq. News.
1983; 43: 236–237.
11.
Chadee DD, Maitre A l, Connell NK:
The collection of Haemagogus equinus Theobald breeding in household containers in Tobago WI.
Mosq. News.
1981; 41: 568–569.
12.
Parra C, Liria J:
Haemagogus equinus Theobald 1903 (Diptera: Culicidae) en el Campus de la Universidad de Carabobo. Valencia. Venezuela.
Salus.
2010; 14: 41–43.
13.
Blohm G, Elbadry MA, Mavian C, et al.:
Mayaro as a Caribbean traveler: Evidence for multiple introductions and transmission of the virus into Haiti.
Int. J. Infect. Dis.
2019; 87: 151–153. PubMed Abstract
| Publisher Full Text
| Free Full Text
14.
Lednicky J, De Rochars VMB, Elbadry M, et al.:
Mayaro virus in child with acute febrile illness, Haiti, 2015.
Emerg. Infect. Dis.
2016; 22: 2000–2002. PubMed Abstract
| Publisher Full Text
| Free Full Text
15.
White SK, Mavian C, Elbadry MA, et al.:
Detection and phylogenetic characterization of arbovirus dual-infections among persons during a chikungunya fever outbreak, Haiti 2014.
PLoS Negl. Trop. Dis.
2018; 12: e0006505. Publisher Full Text
16.
Chan-Chable RJ, Martínez-Arce A, Mis-Avila PC, et al.:
DNA barcodes and evidence of cryptic diversity of anthropophagous mosquitoes in Quintana Roo, Mexico.
Ecol. Evol.
2019; 9: 4692–4705. PubMed Abstract
| Publisher Full Text
| Free Full Text
17.
Gibson CM, Kao RH, Blevins KK, et al.:
Integrative Taxonomy for Continental-Scale Terrestrial Insect Observations.
PLoS One.
2012; 7: e37528. Publisher Full Text
18.
Adeniran AA, Hernández-Triana LM, Ortega-Morales AI, et al.:
Identification of mosquitoes (Diptera: Culicidae) from Mexico State, Mexico using morphology and COI DNA barcoding.
Acta Trop.
2021; 213: 105730. PubMed Abstract
| Publisher Full Text
19.
Viveros-Santos V, Hernández-Triana LM, Ibáñez-Bernal S, et al.:
Integrated Approaches for the Identification of Mosquitoes (Diptera: Culicidae) from the Volcanoes of Central America Physiographic Subprovince of the State of Chiapas, Mexico.
Vector borne and zoonotic diseases (Larchmont, N.Y.).
2022; 22: 120–137. PubMed Abstract
| Publisher Full Text
20.
Hernández-Triana LM, Garza-Hernández JA, Ortega Morales AI, et al.:
An Integrated Molecular Approach to Untangling Host–Vector–Pathogen Interactions in Mosquitoes (Diptera: Culicidae) From Sylvan Communities in Mexico.
Front. Vet. Sci.
2021; 7. PubMed Abstract
| Publisher Full Text
| Free Full Text
21.
Reidenbach KR, Cook S, Bertone MA, et al.:
Phylogenetic analysis and temporal diversification of mosquitoes (Diptera: Culicidae) based on nuclear genes and morphology.
BMC Evol. Biol.
2009; 9: 298. PubMed Abstract
| Publisher Full Text
| Free Full Text
22.
Cameron SL:
Insect mitochondrial genomics: implications for evolution and phylogeny.
Annu. Rev. Entomol.
2014; 59: 95–117. Publisher Full Text
24.
da Silva FS , Cruz ACR, de Almeida Medeiros DB , et al.:
Mitochondrial genome sequencing and phylogeny of Haemagogus albomaculatus, Haemagogus leucocelaenus, Haemagogus spegazzinii, and Haemagogus tropicalis (Diptera: Culicidae).
Sci. Rep.
2020; 10: 16948. PubMed Abstract
| Publisher Full Text
| Free Full Text
25.
Lemos PS, Monteiro HAO, Castro FC, et al.:
Characterization of mitochondrial genome of Haemagogus janthinomys (Diptera: Culicidae).
Mitochondrial DNA Part A.
2017; 28: 50–51. PubMed Abstract
| Publisher Full Text
27.
Ali R, Gebhardt M, Lupiya J, et al.:
The first complete mitochondrional genome of Anopheles gibbinsi using a skimming sequencing approach. [version 1; peer review: 3 approved].
F1000Res.
2024; 13: 553. PubMed Abstract
| Publisher Full Text
| Free Full Text
29.
Bernt M, Donath A, Jühling F, et al.:
MITOS: improved de novo metazoan mitochondrial genome annotation.
Mol. Phylogenet. Evol.
2013; 69: 313–319. PubMed Abstract
| Publisher Full Text
30.
Afgan E, Baker D, Batut B, et al.:
The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update.
Nucleic Acids Res.
2018; 46: W537–W544. PubMed Abstract
| Publisher Full Text
| Free Full Text
31.
Noble SAA, Pierre SA, Sandiford SL, et al.:
Haemagogus equinus mitochondrion, complete genome. [Dataset].
GenBank.
2024.
32.
Johns Hopkins Bloomberg School of Public Health:
Complete mitochondrial genome of Haemagogus equinus. [Dataset].
Bioproject.
2024.
33.
Johns Hopkins Bloomberg School of Public Health:
llumina seq of Haemagogus equinus. [Dataset].
SRA.
2024.
34.
Johns Hopkins Bloomberg School of Public Health:
Invertebrate sample of Haemagogus equinus. [Dataset].
Biosample.
2024.
1
Department of Basic Medical Sciences, Pharmacology and Pharmacy Section, Faculty of Medical Sciences, The University of the West Indies Mona, Kingston, Jamaica 2
Mosquito Control and Research Unit, The University of the West Indies, Mona, Kingston, Jamaica 3
Department of Microbiology, Faculty of Medical Sciences, The University of the West Indies Mona, Kingston, Jamaica 4
Department of Life Sciences, Faculty of Science & Technology, The University of the West Indies Mona, Kingston, Jamaica 5
The W. Harry Feinstone Department of Molecular Microbiology and Immunology, Johns Hopkins Malaria Research Institute, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland, USA
Simone L. Sandiford
Roles:
Conceptualization,
Project Administration,
Resources,
Supervision,
Writing – Original Draft Preparation,
Writing – Review & Editing
Simmoy A. A. Noble
Roles:
Investigation,
Resources,
Writing – Review & Editing
Samra A. Pierre
Roles:
Investigation,
Resources,
Writing – Review & Editing
Douglas E. Norris
Roles:
Funding Acquisition,
Resources,
Writing – Review & Editing
Renee Ali
Roles:
Data Curation,
Formal Analysis,
Methodology,
Resources,
Visualization,
Writing – Original Draft Preparation
SAAN was supported in part by the University at Buffalo Clinical and Translational Science Institute award UL1TR001412 and the Global Infectious Diseases Research Training Program award D43TW010919. DEN and RA were supported in part by funding from the NSF-Accelerator Project D-688: Computing the Biome (2134862), Johns Hopkins Malaria Research Institute and Bloomberg Philanthropies.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Sandiford SL, Noble SAA, Pierre SA et al. The first mitochondrial genome of Haemagogus equinus from Jamaica [version 2; peer review: 2 approved, 1 approved with reservations]. F1000Research 2025, 13:1504 (https://doi.org/10.12688/f1000research.159115.2)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
Makunin A. Reviewer Report For: The first mitochondrial genome of Haemagogus equinus from Jamaica [version 2; peer review: 2 approved, 1 approved with reservations]. F1000Research 2025, 13:1504 (https://doi.org/10.5256/f1000research.184140.r421208)
The manuscript by Sandiford, et al., describes two mitochondrial genomes for arboviral vector mosquito Haemagogus equinus. The dataset is succinctly described and characterised to ensure that the resulting mitochondrial genomes become a valuable resource for future studies. I only have
... Continue reading
The manuscript by Sandiford, et al., describes two mitochondrial genomes for arboviral vector mosquito Haemagogus equinus. The dataset is succinctly described and characterised to ensure that the resulting mitochondrial genomes become a valuable resource for future studies. I only have several minor comments:
- Illumina read length and layout (single or paired end) are not specified
- "manual adjustments of start and stop codons to match reference _Haemagogus_ mitogenomes in the GenBank repository" - if the sequencing effort did not allow to accurately reconstruct start and stop codon, the accuracy of the remaining sequence and subsequent phylogenetic analysis can be compromised
- Selected optimal substitution model for phylogenetic analysis is not specified. Also, it seems that the entire matrix was treated as a single partition, without considering separate models for different codon positions - is it correct?
- COI analysis looks a bit confusing - the coordinates are given for the entire gene, not the ~658bp fragment used for barcoding. "99.59 % comparable to a COI sequence for Haemagogus spegazzinii (YP_010155459)" seemingly indicates protein identity for 510aa segment in YP_010155459, which does not seem relevant for species identification. While this might be out of scope for this particular study (given the complete MT PCG tree looks convincing and Hg. equinus does not seem to be closely related to any other species with complete MT available), I would suggest building a tree based on nucleotide sequence of COI barcode for all Hg. records in BOLD with extracts from two new mitogenomes added - this would complement and verify the PCG tree results with more observations, but smaller resolution.
Are the rationale for sequencing the genome and the species significance clearly described?
Yes
Are the protocols appropriate and is the work technically sound?
Yes
Are sufficient details of the sequencing and extraction, software used, and materials provided to allow replication by others?
Yes
Are the datasets clearly presented in a usable and accessible format, and the assembly and annotation available in an appropriate subject-specific repository?
Yes
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Makunin A. Reviewer Report For: The first mitochondrial genome of Haemagogus equinus from Jamaica [version 2; peer review: 2 approved, 1 approved with reservations]. F1000Research 2025, 13:1504 (https://doi.org/10.5256/f1000research.184140.r421208)
Wilding C. Reviewer Report For: The first mitochondrial genome of Haemagogus equinus from Jamaica [version 2; peer review: 2 approved, 1 approved with reservations]. F1000Research 2025, 13:1504 (https://doi.org/10.5256/f1000research.174799.r349839)
Sandiford et al sequence full mitochondrial genomes from genome skimming data from two Haemagogus equinus specimens collected from Jamaica. They assemble these into a full mtDNA genome containing the expected gene content. Their introduction is well-written and provides much background
... Continue reading
Sandiford et al sequence full mitochondrial genomes from genome skimming data from two Haemagogus equinus specimens collected from Jamaica. They assemble these into a full mtDNA genome containing the expected gene content. Their introduction is well-written and provides much background information and adequate justification for this study. However, I would argue that the statement in the introduction that “Few studies have employed genomic sequencing to investigate the molecular taxonomy and phylogenetic relationships of Hg. equinus mosquitoes” suggests that this paper is going to provide a genome. Whilst it could be argued that the mtDNA is a genome it is somewhat misleading!
In the Methods there is no information on the sequencing undertaken. Whilst the authors say “molecular analysis as described in [27]” this is insufficient – at the very least there should be a brief description of what was done.
The statement “Nine mitogenomes (two sequenced from this study and seven available from GenBank) were used for analysis” should read “Nine mitogenomes (two sequenced from this study and seven available from GenBank) were used for phylogenetic analysis”
In the Results section, Figure 1 has a very poor resolution. This needs altering. Figure 2 has the albomaculatus and tropical groups highlighted, but which group does Hg. leucocelaenus belong? Is this Splendens? Or if not, are there no mitochondrial genomes from this group? I am not particularly clear on the point of Figure 2.
It would make more sense to produce a tree on a wider-scale (including e.g. representative Aedes and Sabethes or, have a tree to show the reader the position of Hg. equinus within the wider Aedini and then have this zoomed in view of Haemagogus, but for those readers unfamiliar with this genus the Figure does not really say anything of use and the text merely says “The Maximum Likelihood phylogenetic tree places it in the Albomaculatus section”. Was this not known before these mitochondrial sequences were generated?
The authors make no comment on whether the gene order seen in this species is as expected given those of closely related species. They should do so.
This study sequenced two specimens with accession numbers PQ_189398 and PQ_189399. How different were they? Some indication of overall genetic distance would be useful.
Are the rationale for sequencing the genome and the species significance clearly described?
Yes
Are the protocols appropriate and is the work technically sound?
Yes
Are sufficient details of the sequencing and extraction, software used, and materials provided to allow replication by others?
No
Are the datasets clearly presented in a usable and accessible format, and the assembly and annotation available in an appropriate subject-specific repository?
Yes
Competing Interests: No competing interests were disclosed.
Reviewer Expertise: Evolutionary genetics
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Wilding C. Reviewer Report For: The first mitochondrial genome of Haemagogus equinus from Jamaica [version 2; peer review: 2 approved, 1 approved with reservations]. F1000Research 2025, 13:1504 (https://doi.org/10.5256/f1000research.174799.r349839)
Simone Sandiford, Department of Basic Medical Sciences, Pharmacology and Pharmacy Section, Faculty of Medical Sciences,, The University of the West Indies Mona, Kingston, Jamaica
11 Sep 2025
Author Response
Thank you for your insightful comments regarding our manuscript. As suggested, we have replaced Figure 1 with a high quality version, enhanced Figure 2, added further details regarding the molecular
...
Continue readingThank you for your insightful comments regarding our manuscript. As suggested, we have replaced Figure 1 with a high quality version, enhanced Figure 2, added further details regarding the molecular analysis to the methods section and stated the similarity between the 2 sequenced specimens in the results.
Thank you for your insightful comments regarding our manuscript. As suggested, we have replaced Figure 1 with a high quality version, enhanced Figure 2, added further details regarding the molecular analysis to the methods section and stated the similarity between the 2 sequenced specimens in the results.
Competing Interests:All authors declare no competing interest.Close
Simone Sandiford, Department of Basic Medical Sciences, Pharmacology and Pharmacy Section, Faculty of Medical Sciences,, The University of the West Indies Mona, Kingston, Jamaica
11 Sep 2025
Author Response
Thank you for your insightful comments regarding our manuscript. As suggested, we have replaced Figure 1 with a high quality version, enhanced Figure 2, added further details regarding the molecular
...
Continue readingThank you for your insightful comments regarding our manuscript. As suggested, we have replaced Figure 1 with a high quality version, enhanced Figure 2, added further details regarding the molecular analysis to the methods section and stated the similarity between the 2 sequenced specimens in the results.
Thank you for your insightful comments regarding our manuscript. As suggested, we have replaced Figure 1 with a high quality version, enhanced Figure 2, added further details regarding the molecular analysis to the methods section and stated the similarity between the 2 sequenced specimens in the results.
Competing Interests:All authors declare no competing interest.Close
Syafruddin D and Setia Asih PB. Reviewer Report For: The first mitochondrial genome of Haemagogus equinus from Jamaica [version 2; peer review: 2 approved, 1 approved with reservations]. F1000Research 2025, 13:1504 (https://doi.org/10.5256/f1000research.174799.r349846)
The manuscript reports the complete mitochondrial genome sequence of Haemagogus equinus from Jamaica. The genome is circular and includes 13 protein-coding genes, 2 ribosomal RNA genes and 22 tRNA genes. As the member of tribe
... Continue reading
General comment:
The manuscript reports the complete mitochondrial genome sequence of Haemagogus equinus from Jamaica. The genome is circular and includes 13 protein-coding genes, 2 ribosomal RNA genes and 22 tRNA genes. As the member of tribe Aedini and vector of Yellow fever, it might be informative to include mitochondrial genome of Aedes aegypti in the phylogenetic tree.
Specific comments:
The map should clearly indicate the position of the 13 protein coding genes, rRNA genes and 22 tRNA genes in the light and heavy strands of mtDNA (Fig. 1).
The authors should include “discussion” section in the manuscript to describe the characteristic of the genes encode by mitochondrial gnome of the Haemogogus equinus in comparison to cosmopolitan species of the tribe Aedini, Aedes aegypti.
Are the rationale for sequencing the genome and the species significance clearly described?
Yes
Are the protocols appropriate and is the work technically sound?
Yes
Are sufficient details of the sequencing and extraction, software used, and materials provided to allow replication by others?
Yes
Are the datasets clearly presented in a usable and accessible format, and the assembly and annotation available in an appropriate subject-specific repository?
Yes
Competing Interests: No competing interests were disclosed.
We confirm that we have read this submission and believe that we have an appropriate level of expertise to confirm that it is of an acceptable scientific standard, however we have significant reservations, as outlined above.
Syafruddin D and Setia Asih PB. Reviewer Report For: The first mitochondrial genome of Haemagogus equinus from Jamaica [version 2; peer review: 2 approved, 1 approved with reservations]. F1000Research 2025, 13:1504 (https://doi.org/10.5256/f1000research.174799.r349846)
Simone Sandiford, Department of Basic Medical Sciences, Pharmacology and Pharmacy Section, Faculty of Medical Sciences,, The University of the West Indies Mona, Kingston, Jamaica
02 Jul 2025
Author Response
Thank you to the reviewers for the feedback provided.
Our article was submitted as a genome note. Based on the F1000Research guidelines genome notes only require an introduction and
...
Continue readingThank you to the reviewers for the feedback provided.
Our article was submitted as a genome note. Based on the F1000Research guidelines genome notes only require an introduction and a detailed protocol in the methods section which discusses the sequencing of the genome. We have fulfilled both requirements. A results section is optional, and we have chosen to provide this also. A discussion section is not needed hence it was not included.
As it relates to the tree, Psorophora ferox was used as an outgroup as it is also a member of tribe Aedini and like Haemagogus equinus, is a forest mosquito.
Figure 1 was constructed similar to other illustrated figures in genome notes which have previously been published and unanimously approved by peer reviewers in F1000Research as indicated below.
Thank you to the reviewers for the feedback provided.
Our article was submitted as a genome note. Based on the F1000Research guidelines genome notes only require an introduction and a detailed protocol in the methods section which discusses the sequencing of the genome. We have fulfilled both requirements. A results section is optional, and we have chosen to provide this also. A discussion section is not needed hence it was not included.
As it relates to the tree, Psorophora ferox was used as an outgroup as it is also a member of tribe Aedini and like Haemagogus equinus, is a forest mosquito.
Figure 1 was constructed similar to other illustrated figures in genome notes which have previously been published and unanimously approved by peer reviewers in F1000Research as indicated below.
Simone Sandiford, Department of Basic Medical Sciences, Pharmacology and Pharmacy Section, Faculty of Medical Sciences,, The University of the West Indies Mona, Kingston, Jamaica
02 Jul 2025
Author Response
Thank you to the reviewers for the feedback provided.
Our article was submitted as a genome note. Based on the F1000Research guidelines genome notes only require an introduction and
...
Continue readingThank you to the reviewers for the feedback provided.
Our article was submitted as a genome note. Based on the F1000Research guidelines genome notes only require an introduction and a detailed protocol in the methods section which discusses the sequencing of the genome. We have fulfilled both requirements. A results section is optional, and we have chosen to provide this also. A discussion section is not needed hence it was not included.
As it relates to the tree, Psorophora ferox was used as an outgroup as it is also a member of tribe Aedini and like Haemagogus equinus, is a forest mosquito.
Figure 1 was constructed similar to other illustrated figures in genome notes which have previously been published and unanimously approved by peer reviewers in F1000Research as indicated below.
Thank you to the reviewers for the feedback provided.
Our article was submitted as a genome note. Based on the F1000Research guidelines genome notes only require an introduction and a detailed protocol in the methods section which discusses the sequencing of the genome. We have fulfilled both requirements. A results section is optional, and we have chosen to provide this also. A discussion section is not needed hence it was not included.
As it relates to the tree, Psorophora ferox was used as an outgroup as it is also a member of tribe Aedini and like Haemagogus equinus, is a forest mosquito.
Figure 1 was constructed similar to other illustrated figures in genome notes which have previously been published and unanimously approved by peer reviewers in F1000Research as indicated below.
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)