Keywords
Pseudomonas aeruginosa, BTEX, genes rhlABC, genes mlaABCD, genes pqs
This article is included in the Genomics and Genetics gateway.
Pseudomonas aeruginosa, BTEX, genes rhlABC, genes mlaABCD, genes pqs
The edition was refined based on the reviewers' advice to enhance readability and accuracy. The tirle has been ammended. BTEX was defined upon its first mention, and the importance of the study on Pseudomonas aeruginosa for bioremediation was emphasized. The methods section was further detailed, specifying bioinformatics tools, parameters, and selection criteria for reference genomes to ensure reproducibility. In the results section, data presentation was separated from interpretation, and figures and tables were improved with clearer annotations. In the discussion, the genetic organization of the rhl and mla genes was examined in greater depth, comparing it with previous studies and highlighting its impact on hydrocarbon tolerance. The potential applications in bioremediation were also emphasized. However, a discussion on accessory genes was not included, as it is not the focus of the study. Finally, the conclusion was expanded to summarize key findings and suggest future research directions on gene-environment interactions in hydrocarbon degradation. References were reviewed to ensure consistency and the inclusion of relevant studies.
See the authors' detailed response to the review by Andi Kurniawan
Polycyclic aromatic hydrocarbons (PAHs) and mono aromatic hydrocarbons BTEX (benzene, toluene, ethylbenzene, and xylene), are widely distributed in the environment as complex mixtures. They present a significant environmental challenge due to their abundance and global recognition as pollutants (Andreoni & Gianfreda, 2007). The World Health Organization has classified BTEX as hazardous compounds, with benzene being particularly concerning due to its status as a Group 1 carcinogen (Chicca et al., 2020). However, other BTEX compounds also pose health risks. Prolonged exposure to toluene and xylene can negatively impact the respiratory and central nervous systems, leading to conditions such as asthma, shortness of breath, persistent coughing, wheezing, and chest tightness, along with neurological effects like headaches, dizziness, nausea, fatigue, agitation, and confusion (Barros et al., 2019). Long-term exposure to ethylbenzene has also been associated with kidney damage and an increased risk of cancer (ATSDR, 2010). Additionally, BTEX compounds not only threaten human health but also play a significant role in atmospheric pollution. Their interaction with nitrogen oxides (NOx) contributes to the formation of ground-level ozone (O3), which exacerbates air quality issues and poses further environmental risks (Golkhorshidi et al., 2019). Petroleum hydrocarbon spills further compound these issues, as they act as a major source of soil and water contamination, disrupting both aquatic and terrestrial ecosystems by affecting their structure and biological processes (Shi et al., 2015). Due to their persistence in the environment, these pollutants cause harm to human health, flora, and fauna, making them a critical focus of environmental and public health studies (Dongen, 2000).
In spite of the toxicity risks, hydrocarbon polluted places are the best source to isolate aromatic and aliphatic hydrocarbon degrading microoganisms (Salgado Brito et al., 2008). Among these microorganisms are bacteria belonging to various genera such as Bacillus, Burkholderia, Acinetobacter, Rhodococcus, and Pseudomonas (Ghosal et al., 2016). For instance, strains like Pseudomonas aeruginosa N002, P. aeruginosa SJTD-1, and P. aeruginosa DQ8 have been identified as alkane-degrading organisms, while Massilia aromaticivorans, Pseudomonas putida, Pseudomonas fluorescens, Rhodococcus, Bacillus sp., and Streptomyces sp. have shown potential for BTEX degradation (Kaur et al., 2023). Additionally, Acinetobacter spp., Alcaligenes spp., Acidovorax spp., Janibacter spp., and Rhodococcus spp. have been reported to be capable of degrading PAHs (Shahsavari et al., 2019).
Studies related to the characterization and annotation of genes involved in hydrocarbon degradation are useful to understand the physiological and metabolic processes in the intrinsic dynamics of native microorganisms at the level of molecular biodiversity, genomics, transcriptomics, and metabolomics (Kumari et al., 2023).
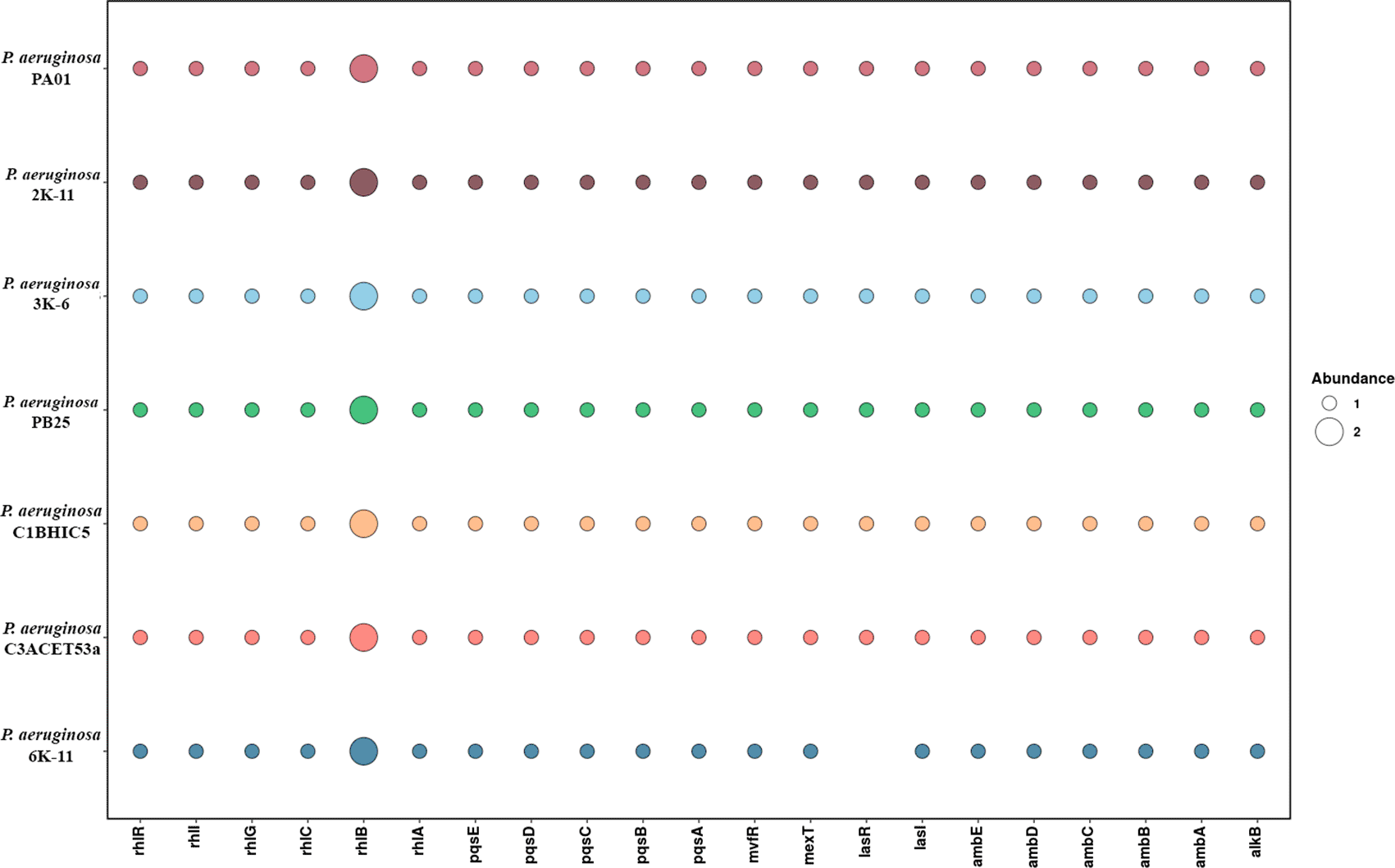
In Pseudomonas aeruginosa, it is necessary to consider the quorum sensing hierarchical complex network (García-Reyes et al., 2020; Kuang et al., 2020; J. Lee & Zhang, 2015; Meng et al., 2020): the rhl system (rhlABR operon, rhlC, rhlI, and rhlG genes); las system (lasR, lasI genes) (Kuang et al., 2020; Meng et al., 2020; O’Connor et al., 2022); PQS system (pqsABCBDE operon, pqsH, pqsL, and pqsR or mvfR genes) (Meng et al., 2020; O’Connor et al., 2022; Xiao et al., 2006); and the IQS system (ambBCDE operon, iqsR gene) (J. Lee & Zhang, 2015; O’Connor et al., 2022). Additionally, genes responsible for BTEX tolerance (mlaABCD genes), as well as those involved in alkane degradation (alkB gene) (Cruz et al., 2020), BTEX (tod, tom, tbu, tou, pTOL pathways), and PAHs (TOD, NAH, OCT plasmid genes, among others), have been reported.
Studies of genes related to the degradation of monoaromatic hydrocarbons have been carried in Pseudomonas putida. The toluene-degrading plasmid (TOL) has been studied in P. putida mt-2, which encodes metabolic pathways for the degradation of toluene, m-xylene, and p-xylene into carboxylic acids. The xylN genes of the TOL plasmid encode a m- xylene transporter porin (Kasai et al., 2001). Similarly, the degradation of toluene follows six differentiated metabolic pathways, which involve the tod gene encoding toluene dioxygenase, the tmo/tbm/tbc genes responsible for synthesizing toluene 2-monooxygenase enzyme, tbu/tbh genes encoding toluene-3-monooxygenase, tmoABCDEF genes encoding different subunits of ortho toluene 4-monooxygenase protein, and tbc1FEDCBA/tbc2ABCDEF genes encoding meta toluene 4-monooxygenase (Parales et al., 2008). On the other hand, the genes involved in the aerobic degradation of benzene are the benzA genes encoding an enzyme that carries out the initial ring cleavage oxidation reaction of monoaromatic hydrocarbons, and this enzyme has been studied in Pseudomonas putida AQ8 (Chicca et al., 2020). To date, there are not many records involving strains of the species Pseudomonas aeruginosa in the degradation of monoaromatic hydrocarbons, nor annotated genes associated with this process.
Research on hydrocarbon degradation has extensively explored various species of Pseudomonas, particularly focusing on their ability to break down complex organic compounds in diverse environments. Pseudomonas aeruginosa, renowned for its fluoranthene degradation capability, exhibits robust metabolic capacities. Investigations on this species have spanned multiple domains, including genomics, transcriptomics, metabolomics, and its potential applications in biotechnology (He et al., 2018).
This research is focused in comparing the genetic characterization of genes coding for rhamnolipids production (rhl ) and BTEX degradating genes in six Pseudomonas aeruginosa strains isolated in hydrocarbon polluted environments.
Six strains of Pseudomonas aeruginosa isolated from hydrocarbon contaminated environments, from different geographical sites in Peru (Figure 1) were selected ( Table 1); belonging to the collection of microorganisms of the Laboratory of Microbiology and Microbial Biotechnology (LAMYBIM) of the Faculty of Biological Sciences of the U.N.M.S.M. (Palomino et al., 2017).
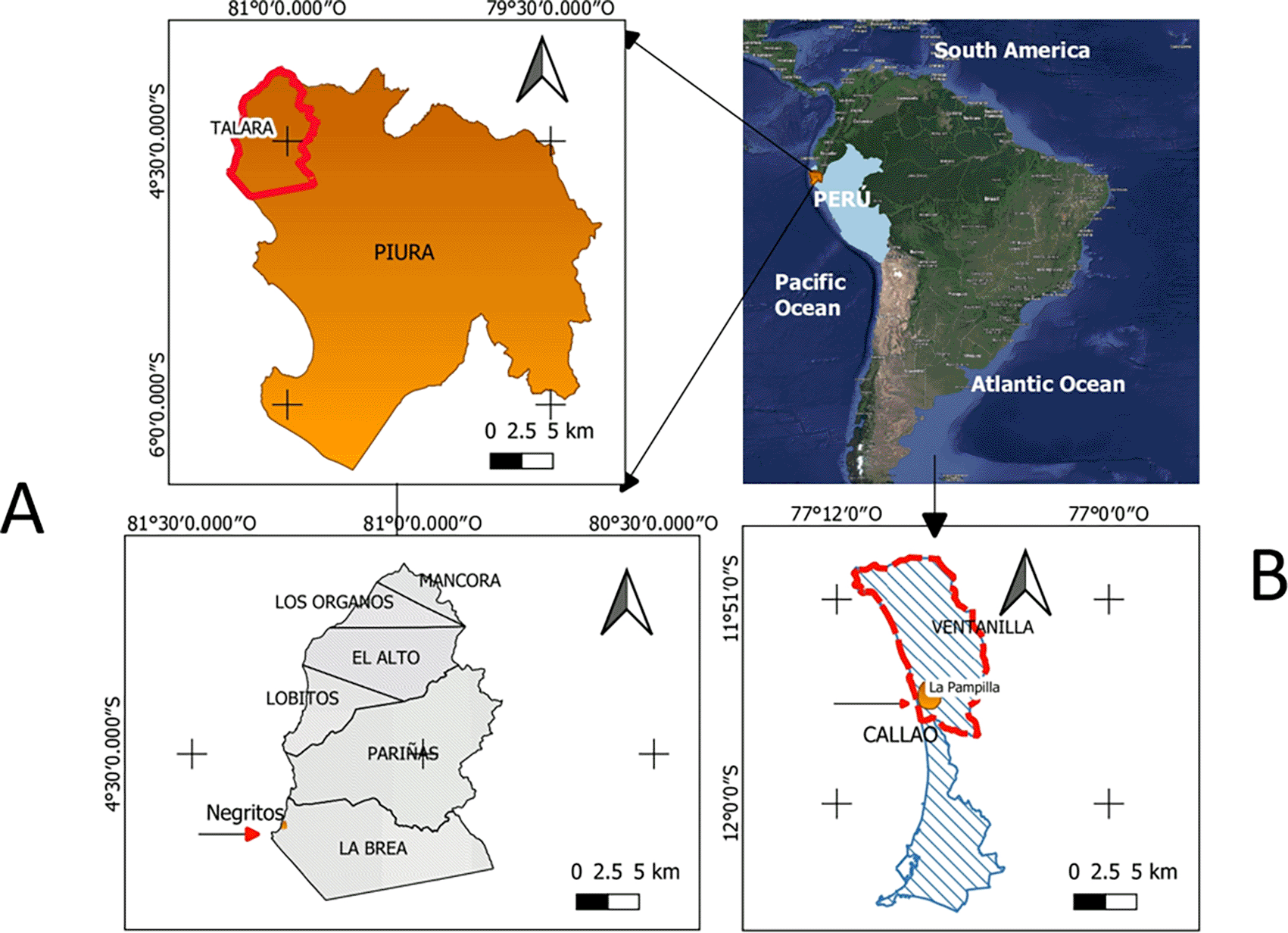
Elaboration: Figure created by the authors as part of this study.
Columns describe the selection criteria for the strains, the names, the place of sampling, the geographic location and the reference.
| Selected strains | Codes | Sampling location | Geographic location | Reference |
|---|---|---|---|---|
| Rhmanolipid hyperproducer | 6K-11 | Environments contaminated with hydrocarbons | Talara, Northern Peru | Tabuchi et al., 2015 |
| With emulsifying capacity and heavy metal removal efficiency | PB25 | Environments contaminated with hydrocarbons | La Pampilla, Ventanilla | Giraldo et al., 2014 |
| With Hydrocarbonoclastic capacity | C1BHIC5 | Land treatment field contaminated with crude oil | Talara, Northern Peru | Terán Morales, 2020 |
| With Hydrocarbonoclastic capacity | C3ACETC53a | Land treatment field contaminated with crude oil | Talara, Northern Peru | Terán Morales, 2020 |
| Non-rhamnolipid producer | 2K-1 | Environments contaminated with hydrocarbons | Talara, Northern Peru | Palomino et al., 2017 |
| Non-rhamnolipid producer | 3K-6 | Environments contaminated with hydrocarbons | Talara, Northern Peru | Palomino et al., 2017 |
For the purification process, the strains were subcultured in glass flasks containing 50 mL of Luria Bertani medium, at 37 °C overnight. Subsequently, the methodology of the innuPREP Bacteria DNA kit from Analytik Jena was followed.
Raw reads were verified with FastQC software (Andrews, S., 2010) and filtered to remove contaminating Illumina adapter sequences and quality trimmed with Trimmomatic v0.36 (Salgado Brito et al., 2008). The resulting filtered reads were assembled using SPAdes v3.14.1 (Ghosal et al., 2016). Assemblies were then filtered to contain only contigs longer than 400 bp. The quality of the assemblies was checked with Quast (Ivanova et al., 2022). For validation assembly, we used Busco (Khan et al., 2001) and mapped reads filtered against the draft genome using Bowtie2 v2.5.4 (Zylstra & Gibson, 1989) and Samtools v1.21 (Furukawa et al., 1993). For annotation, we used the Prokka v1.14.6 pipeline, which used Prodigal v2.6.3 (Harayama & Rekik, 1993), RNAmmer v1.2 (Reineke, 1998), Aragorn v1.2.36 (Mapelli et al., 2017), and MinCED v0.4.2 (Skennerton et al., 2021) to find protein-coding genes, RNA, tRNA, and CRISPR regions, respectively. Finally, we found groups of orthologs using Orthofinder v3.0.1.b1 (Cao et al., 2015), adding the proteome of a Pseudomonas aeruginosa strain (GCA_000006765.1) for this analysis.
Trimming quality (Phred Q > 25) and removal of adapters were conducted with Trimmomatic v0.36 (Bolger et al., 2014) and TrimGalore v0.6.10 software (Felix Krueger et al., 2023), respectively. De novo assembly was performed using SPAdes v3.10.1 (Bankevich et al., 2012), testing different k-mers (from 23 to 123). SSPACE v2.0 (Boetzer et al., 2011) was used to join contigs to build scaffolds with an iteration of raw read alignment over the contigs to minimize the gaps. A final gap-filling step was performed using GapCloser v.1.2.1 (Boetzer & Pirovano, 2012) to generate a draft genome. We used QUAST v5.2.0 (Gurevich et al., 2013) for assembly statistics. The completeness and consistency of the assembled genome were estimated using the Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.8.2 software (Simão et al., 2015) and CheckM (Parks et al., 2015). These assemblies have been deposited at DDBJ/ENA/GenBank under the accessions: JAOAQG000000000, JAOAQH000000000, JAOAQI000000000, JAOAQJ000000000, JANATH000000000, JANZYL000000000.
The functional annotation of the obtained genomes was carried out using BAKTA v1.6.1 (Schwengers et al., 2021), a precise annotation tool, using a local conda environment (Anaconda v23.5.0). We used BAKTA’s database in its 2022-08-25 release. Standard parameters were used for this functional annotation.
For comparative analyses, high-quality Pseudomonas aeruginosa genomes were retrieved from public repositories, specifically from the Reference Sequences Database NCBI (RefSeq; October 2022). The selection criteria were based on genome completeness, assembly quality, and annotation accuracy, as assessed using QUAST v5.2.0 (Gurevich et al., 2013). Only genomes classified as “complete” or “high-quality draft” were included, ensuring reliable comparisons with the newly sequenced strains.
Average Nucleotide Identity (ANI) analysis was performed using pyANI (v. 0.2.12) (Pritchard et al., 2016). A heatmap was generated using the pheatmap package in R (v. 4.1.2). A tblastx was performed using the assembled genomes with a focus in 2 genes of interest, and the genetic diagram was built using the genomes package in R. The phylogenetic reconstruction of multiple Pseudomonas aeruginosa strains was built, leveraging the core gene alignment obtained during pangenome reconstruction through Panaroo (Tonkin-Hill et al., 2020). Subsequently, RAxML v8.2.13 (Stamatakis, 2014) was employed to construct a phylogenetic tree based on the aligned core genes. The model was selected using ModelTest NG (Darriba et al., 2020), with the TVM+I+G4 model chosen as the optimal one for this analysis. The phylogenetic analysis was run with 100 bootstraps. The phylogenetic tree was graphically edited using the iTOL v5 web tool (Letunic & Bork, 2021).
For gene topology and synteny, we performed a tblastx between the contigs of interest. We performed this for 2 genes of interest: rhl and mla. For drawing the topology and the synteny obtained, we used the R package, gggenome (v1.0.1) (Hackl T. et al., 2024). For the rhl system, we filtered the tblastx results based on E-value (<0.01), coverage length (>=20) and identity (>=30). For the mla system, we filtered by criteria based on E-value (<0.01), coverage length (>=100) and identity (>=30). PB25’s alignment against P. putida was filtered following more lax criteria (E-value (<0.01), coverage length (>=20) and identity (>=30)).
The assembled genomes had lengths ranging from approximately 5.6 Mbp to 6 Mbp. The 2K-1 strain had the longest genome at approximately 6 Mbp. The strains C3ACET53a and C1BHIC5 had genome lengths of 5.88 Mbp and 5.84 Mbp, respectively, while the remaining strains had genome lengths close to 5.6 Mbp. The GC content was consistent across all strains, with values around 66%. The N50 values varied among the genomes, with the highest N50 observed in strain C1BHIC5 (Table 2).
The Average Nucleotide Identity (ANI) was assessed among the sequenced Pseudomonas aeruginosa strains using the MUMMER algorithm. All strains showed ANI values above 0.95, confirming their classification within the Pseudomonas aeruginosa species (Jain et al., 2018). The ANI values ranged from 97.5% to 99.9% (Figure 2).
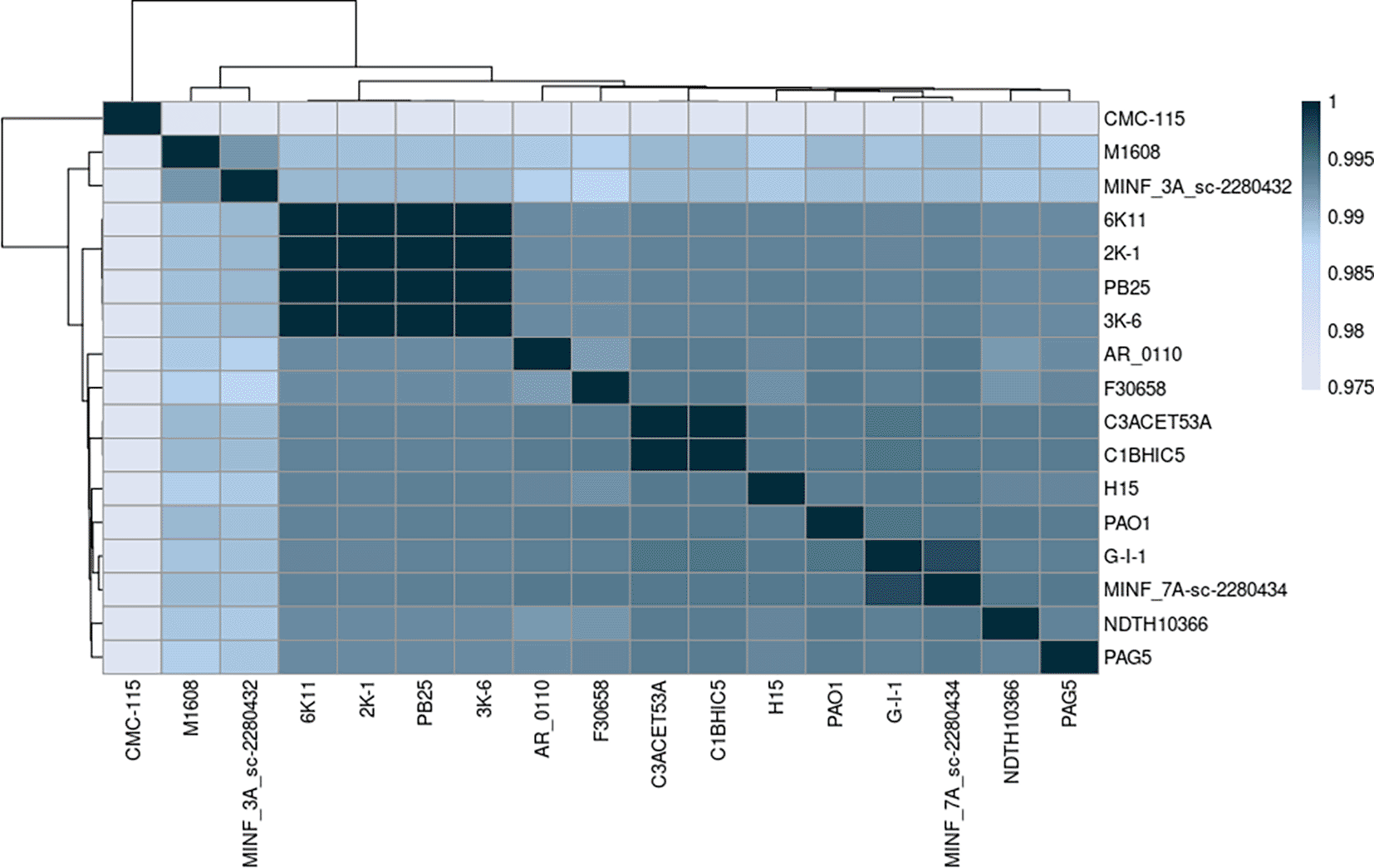
Elaboration: Figure created by the authors as part of this study.
The pangenome analysis, shown in Figure 3A, includes a comparison between the isolated strains, PAO1, and PA7. Among the eight analyzed strains, 4,068 genes were identified as shared. Of these, 65 genes were common to all isolated strains, excluding PA7 and PAO1. Additionally, 108 genes were exclusively shared by C3ACET53A and C1BHIC5, while 197 genes were exclusively shared by the group comprising 6K-11, PB25, 3K-6, and 2K-1.
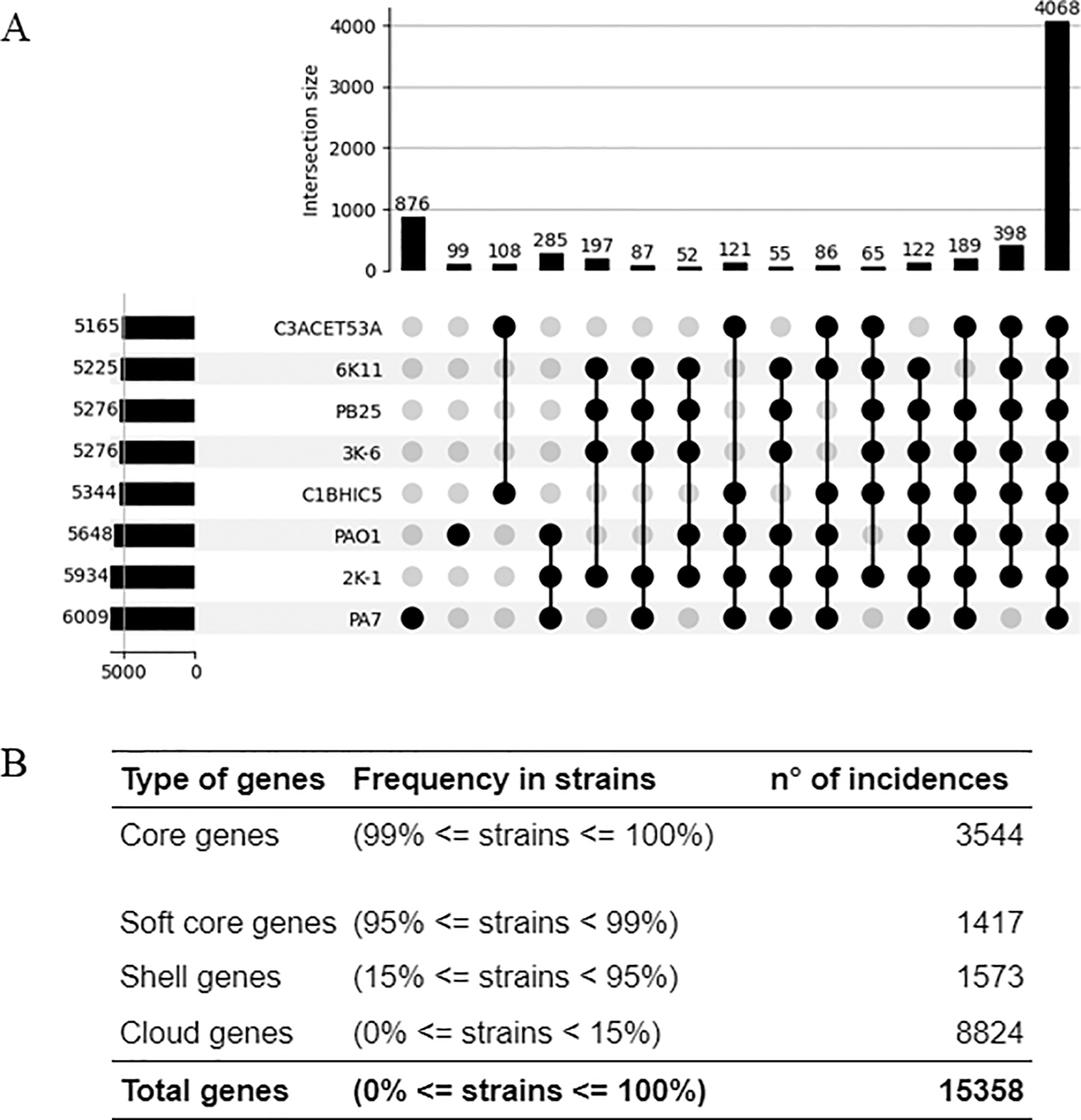
Elaboration: Figure created by the authors as part of this study.
A total of 3,544 genes were classified as core genes in a pangenome constructed with 93 strains, including the newly sequenced isolates. In contrast, 11,814 genes were identified as accessory or variable genes, classified into three categories: softcore, shell, and cloud genes, based on their presence across the genomes (Figure 3B).
A phylogenetic tree was constructed based on various strains previously reported in other studies. Bootstrap values reached 100 in most cases. The branch length for PA7 was adjusted due to its high divergence (original branch length = 9.9999513099). We attempted to reconstruct the Pseudomonas aeruginosa “biotypes” found by Palleroni et al. (1973). The analysis placed all six sequenced strains within Clade 1, which also includes the reference strain PAO1. Within this clade, the strains were grouped into two distinct clusters. The strains 6K-11, 2K-1, PB25, and 3K-6 clustered with strains T38079, F9670, S86968, ATCC 27853, PA96, and M18, while strains C1BHIC5 and C3ACET53A formed a separate cluster with strains 8380 and SJTD-1 (Figure 4).
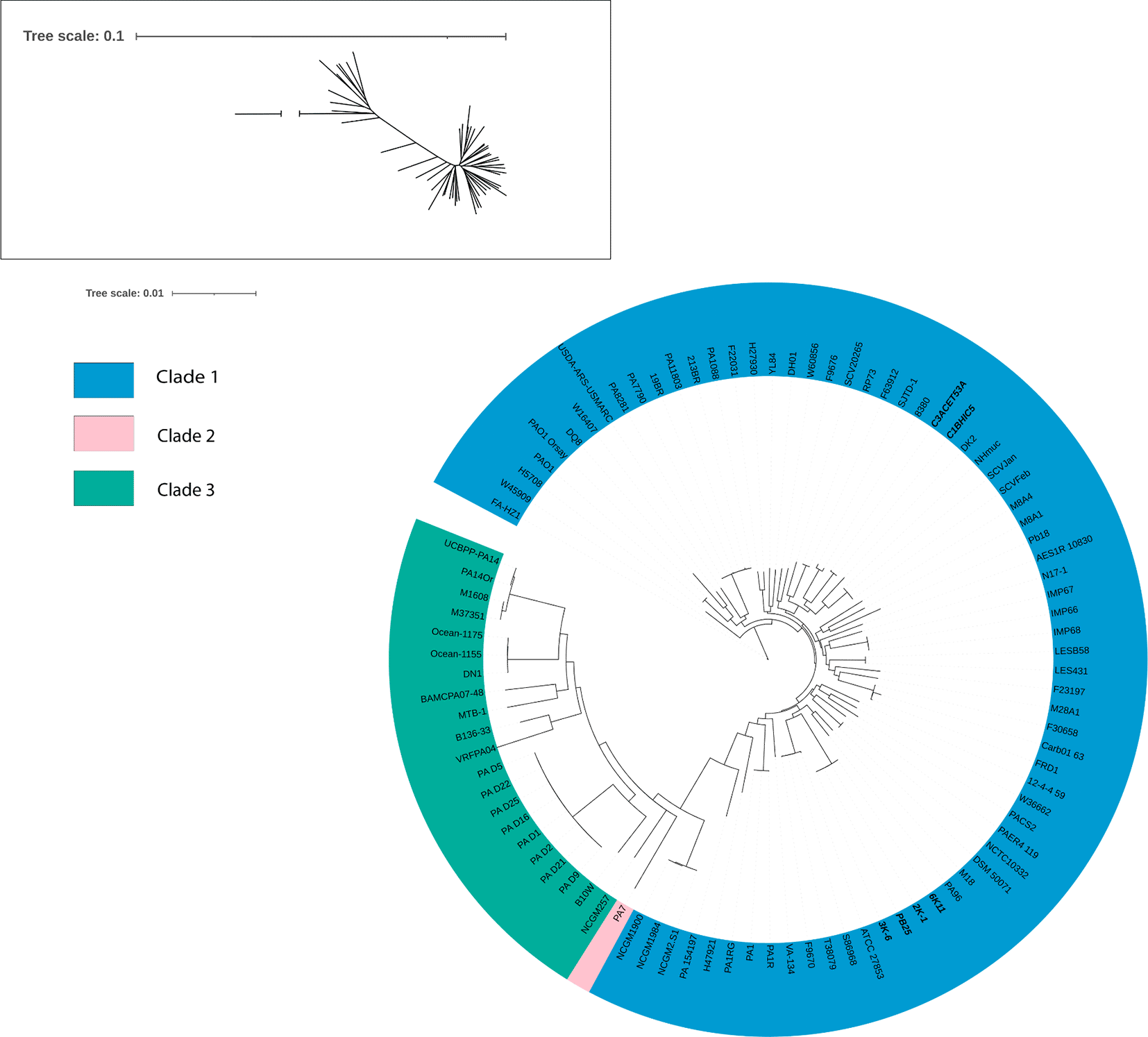
Bootstrap values reached a value of 100 for most cases. PA7’s branch length had to be reduced because of its unusual length because of its divergence (branch length = 9.9999513099). In bold letters, the isolates of interest.
Elaboration: Figure created by the authors as part of this study.
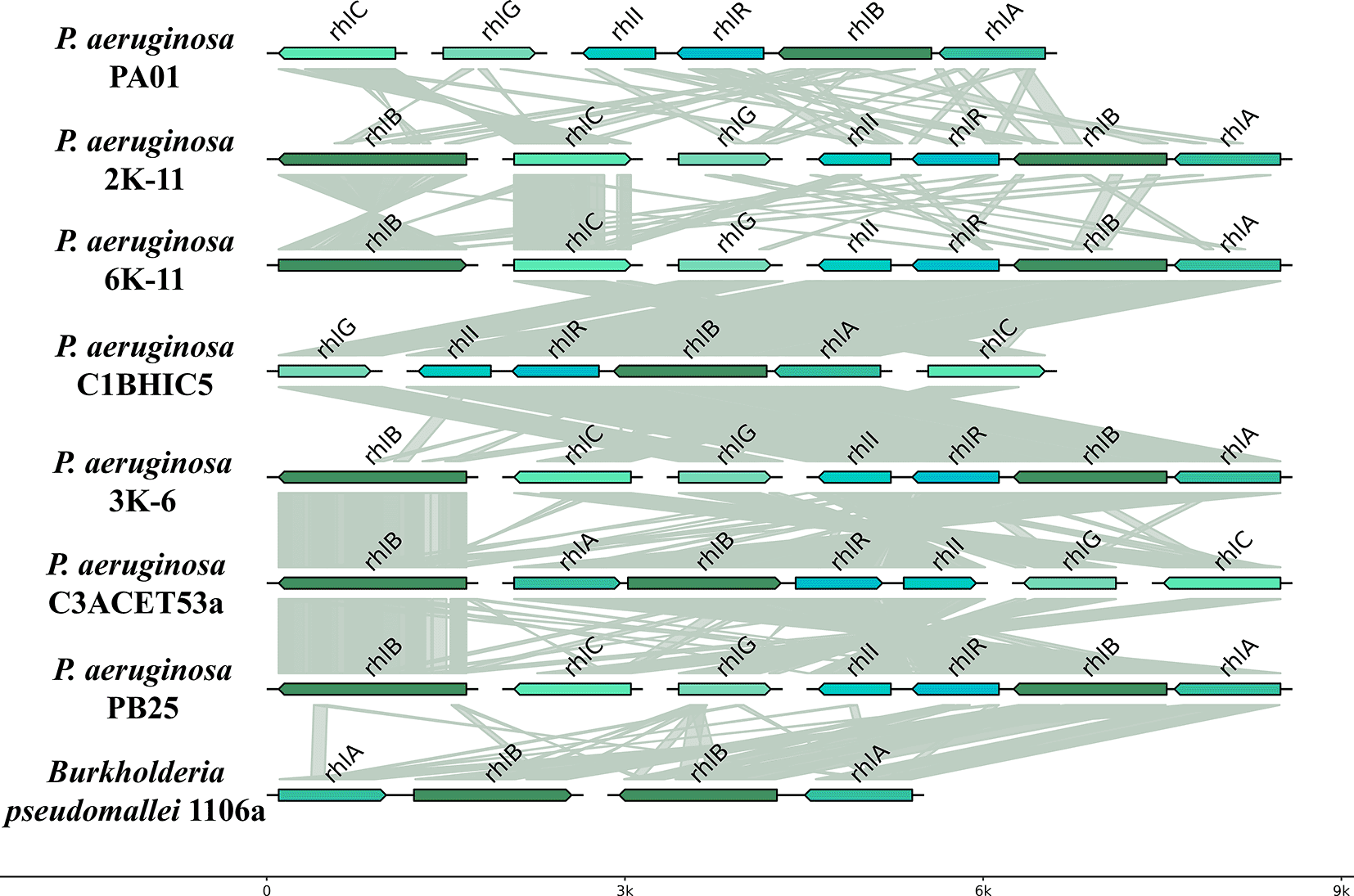
The organization and synteny of rhl genes were evaluated across several bacterial strains. All analyzed strains contained a duplicated copy of the rhlB gene, except for strain C1BHIC5, which had a single copy. The rhl genes maintained a consistent organization across strains, with the rhlABRI genes always positioned adjacent to each other. In most strains, this cluster was arranged in a 3′-5′ orientation, except for strain C3ACET53A. The rhlG gene was consistently located near the rhlC gene, but its position varied across strains. The rhlG gene was oriented in a 5′-3′ direction in all strains except for C3ACET53A. Similarly, rhlC exhibited positional variability but remained adjacent to rhlG. The rhlB gene appeared in two locations within the genome: one copy within the rhlABRI cluster and another in a separate genomic region. The rhlABRI cluster was also present in the PAO1 genome.
Synteny analysis revealed high conservation of the rhlB gene among the recovered isolates. The copy located outside the rhlABRI cluster exhibited strong synteny across all strains (Table 3).
The presence and organization of the mla system were also examined (Figure 7). All sequenced Pseudomonas aeruginosa strains exhibited a similar genomic structure for this system. Most strains contained two copies of the mlaA gene, except for strain C1BHIC5, which had a single copy. The mlaFEDC cluster was identified in all strains, whereas the mlaEFD cluster was absent in strain C3ACET53A. The orientation of these clusters varied among strains. Synteny analysis indicated high similarity between these clusters in the sequenced strains. Comparison with Pseudomonas putida showed that the mla genes were located between the murA and ppcD genes, a genomic arrangement similar to that observed in other P. aeruginosa strains. Additionally, the ttg2D gene, previously associated with toluene tolerance, was found in the mla gene region of P. putida.
Elaboration: Figure created by the authors as part of this study.

Elaboration: Figure created by the authors as part of this study.
Elaboration: Figure created by the authors as part of this study.
Several studies seek to evaluate the bioremediation potential of Pseudomonas aeruginosa against hydrocarbons, such as Baig et al. (2022), who isolated 2 strains: BAA-427 and ATCC-27853, from oil-contaminated soils (Aslam Refinery, Pakistan); Liu et al. (2022) also isolated P. aeruginosa AQNU-1 in water samples from a lake wetland near a petrochemical industry (Anqing City, China) and Mahjoubi et al. (2021) isolated a halotolerant strain of P. aeruginosa UN14 in hydrocarbon-contaminated sediment (refinery on the coast of the port of Bizerte, northern Tunisia). In Peru, the Laboratory of Microbiology and Microbial Biotechnology of the U.N.M.S.M. has actively isolated strains of Pseudomonas aeruginosa from polluted environments, which present efficient physiological capacity in relation to hydrocarbon degradation. Of interest are strains with hyper-producing capacity of rhamnolipids (Pseudomonas aeruginosa 6K-11); with emulsifying capacity and efficiency for the removal of heavy metals (Pseudomonas aeruginosa PB25) and with hydrocarbonoclastic activity (Pseudomonas aeruginosa C1BHIC5 and Pseudomonas aeruginosa C3ACETC53a) (Palomino, Roger A et al., 2017). These strains present a high potential for hydrocarbon bioremediation through biosurfactant production, emulsification, and degradation of toxic compounds such as BTEX. Synthetic biology enhances these processes by optimizing metabolic pathways, engineering microbial consortia, and developing biosensors for real-time contaminant monitoring (Jiménez-Díaz et al., 2022; Bhattacharjee et al., 2020). However, regulating catabolic gene expression remains a challenge, as hydrocarbon degradation systems vary even among closely related species. Research has predominantly focused on alkane-sensing systems for C5-C18 compounds and AlkB or CYP enzymes, overlooking metabolic pathways for smaller hydrocarbons, particularly those linked to SDIMOs and CuMMOs. Addressing these gaps could improve biosensor design and facilitate regulatory approvals for field applications (Moratti et al., 2022).
The species phylogeny was constructed using strains previously reported in various studies, focusing especially on the “biotypes” identified by Palleroni et al. (1973). The branch length for PA7 had to be reduced because of its unusual divergence (original branch length = 9.9999513099). Our strains, originated from environmental sources, formed 2 distinct groups within Clade 1, along with clinical origin strains. Unlike other pathogenic bacteria, genomic studies have shown that clinical and environmental isolates of P. aeruginosa do not exhibit particular genomic differences, confirming that genetic variability related to the environment is reduced in this species (Grosso-Becerra et al., 2014). This would explain why the origin of the strains did not determine the clade they formed in the phylogeny of P. aeruginosa. Likewise, it would also explain the results observed in the ANI matrix, in which the percentages are not very different from each other.
The pangenomic comparison also showed a large number of accessory genes (n=10,397; 67%) and a reduced core genome (n=3,544; 23%). This coincides with previous pangenomic studies that have highlighted the importance of horizontal gene transfer in this species (Freschi et al., 2019).
Pseudomonas aeruginosa utilizes a quorum sensing (QS) system to regulate the synthesis of rhamnolipids (RLs), which are essential for the emulsification and assimilation of hydrocarbons (Xu et al., 2020). The rhlABC genes are responsible for the production of these RLs in Pseudomonas aeruginosa strains isolated from petroleum-contaminated environments (Castro et al., 2022; Ji et al., 2016). These genes encode the enzymes RhlA, RhlB, and RhlC, which transform precursors into mono-rhamnolipid and di-rhamnolipid. The rhl gene family, primarily found in Pseudomonas aeruginosa and Burkholderia species, is responsible for the biosynthesis of rhamnolipids. We evaluated the organization and synteny of rhl genes in several bacterial strains. Our strains presented a double copy of the rhlB gene, except for the C1BHIC5 strain, which had only one copy. The rhl genes did not exhibit a radically different organization across all evaluated strains. The rhlABRI cluster is also part of the PAO1 genome. Furthermore, we observed significant synteny between the rhlB gene in the recovered isolates. The copy situated far from the rhlABRI presents a clear synteny among all strains (Table 3). We have evidenced the presence of these genes in all strains analyzed through comparative genomic studies ( Figure 5).
Regarding quorum sensing interactions, the las system regulates the expression of virulence genes and activates the pqs and rhl systems (Lee & Zhang, 2015), while the pqs system regulates more than 35 loci related to virulence (Li et al., 2022). The rhl system controls both virulence genes and rhamnolipid production (García-Reyes et al., 2020). In our analysis of these genes across several strains, we found that all strains have a similar gene abundance. However, the 6K-11 strain stands out due to the absence of the lasR gene (Figure 7). These findings regarding the absence or presence of these genes could be due to the respective virulence mechanisms in each strain (Dötsch et al., 2015).
In terms of the degradation of BTEX hydrocarbons (benzene, toluene, ethylbenzene, and xylene), specific genes responsible for this process have already been identified in various microorganisms (Choi et al., 2013; Lee et al., 2019; Lin et al., 2002), primarily studied in species of Pseudomonas and P. putida (Bacosa et al., 2021). In our study, in an attempt to corroborate hydrocarbonoclastic physiological activity, we searched for these genes within the complete genome but were unsuccessful in all the strains analyzed. Therefore, we searched for orthologous genes from the ttg2 operon previously found in Pseudomonas putida, initially linked to toluene tolerance (Yero et al., 2021), with this system being the mla. MlaA removes glycerophospholipids from the outer leaflet of the outer membrane (OM) and deliver them to the MlaFEDB complex in the inner membrane (IM) via the periplasmic substrate-binding protein MlaC (Kaur et al., 2023).
The genetic organization of the mla system in the synteny showed discontinuity, as the mlaCDEF genes are grouped in an operon, while mlaA is separated in the genome. This aligns with what was reported by Kaur and Mingeot-Leclercq (2024), who mentioned that in gram-negative bacteria, including P. aeruginosa, the system is organized in this way. However, the mlaB gene was not found in the operon, consistent with the absence of this gene in alpha-proteobacteria and epsilon-proteobacteria. Given the toluene tolerance previously shown by the strains (Querebalú García, 2020, data not available), the mla system was sought as a gene operon of interest. Genomically, this system was present with a similar structure in all the isolated P. aeruginosa strains (Figure 7). Almost all strains exhibited two copies of the mlaA gene, except for the strain C1BHIC5, which presented only one copy. The mlaFEDC cluster was present in all strains, whereas the mlaEFD cluster was present in all strains except for C3ACET53A. The mla gene blocks were highly similar between the analyzed strains. When compared with Pseudomonas putida, this species presented the mla genes between the murA and ppcD genes, in a similar conformation to other P. aeruginosa strains. Notably, the gene responsible for toluene tolerance, ttg2D, was also found among these genes in P. putida.
Benzene, toluene, ethylbenzene, and xylene, collectively known as BTEX, are common pollutants of soils and groundwater. BTEX and other polycyclic aromatic hydrocarbons are extremely toxic to microorganisms due to their accumulation in hydrophobic cell membranes (Choi et al., 2013; Hąc-Wydro et al., 2019). Therefore, the genes studied may be involved in resistance to BTEX. Pseudomonas aeruginosa possesses a complex genetic and enzymatic regulatory system that allows it not only to produce rhamnolipids essential for the emulsification and assimilation of hydrocarbons but also to tolerate aromatic compounds such as BTEX.
This study provides a comprehensive genomic characterization of six Peruvian Pseudomonas aeruginosa strains isolated from hydrocarbon-contaminated environments. The analysis revealed key genetic determinants associated with rhamnolipid (rhl genes) biosynthesis, BTEX degradation, and membrane lipid homeostasis (mla genes), highlighting their potential for bioremediation applications. Comparative genomics demonstrated that these strains cluster within Clade 1 of P. aeruginosa and possess a conserved yet variable genetic architecture for hydrocarbon degradation, stress tolerance mechanisms, and lipid transport systems.
Studies on genes related to the degradation of monoaromatic hydrocarbons have been conducted in Pseudomonas putida. The toluene-degrading plasmid (TOL) has been studied in P. putida mt-2, which encodes metabolic pathways for the degradation of toluene, m-xylene, and p-xylene into carboxylic acids. The xylN genes of the TOL plasmid encode a porin transporter for m-xylene (Kasai et al., 2001). Similarly, toluene degradation follows six differentiated metabolic pathways, involving the tod gene, which encodes the enzyme toluene dioxygenase; the tmo/tbm/tbc genes responsible for the synthesis of the Toluene 2-monooxygenase enzyme; tbu/tbh genes that encode Toluene 3-monooxygenase; tmoABCDEF genes that encode different subunits of the ortho Toluene 4-monooxygenase protein; and tbc1FEDCBA/tbc2ABCDEF genes that encode meta Toluene 4-monooxygenase (Parales et al., 2008). On the other hand, the genes involved in the aerobic degradation of benzene are the benzA genes, which encode an enzyme that carries out the initial oxidation reaction of the ring-cleavage of monoaromatic hydrocarbons, and this enzyme has been studied in Pseudomonas putida AQ8 (Chicca et al., 2020). To date, there are not many records involving strains of the species Pseudomonas aeruginosa in the degradation of monoaromatic hydrocarbons, nor annotated genes associated with this process.
These findings contribute to the growing body of knowledge on microbial genomics and bioremediation by identifying novel genetic components involved in hydrocarbon degradation, membrane transport, and rhamnolipid-mediated emulsification. Understanding the genetic basis of these metabolic pathways not only enhances our ability to harness microbial communities for environmental restoration but also opens avenues for synthetic biology applications aimed at optimizing bioremediation efficiency.
Future research should explore gene-environment and accesory genes interactions in greater depth, particularly the regulatory mechanisms governing hydrocarbon degradation pathways and lipid transport systems. Additionally, metagenomic and transcriptomic studies could provide insights into microbial community dynamics in contaminated sites, further refining bioremediation strategies.
If you are using Overleaf, either select “Submit” then F1000Research, or click “Submit to F1000Research” in the top right-hand corner. Alternatively, generate a PDF file of your project and submit this alongside a zip file containing all project files (includes the source files, style files, and PDF) using online submission form.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)