Keywords
Neuroendocrine Tumor, Duodenal NET, Humoral Hypercalcemia of Malignancy, Parathyroid Hormone Related Protein, Paraneoplastic Syndromes, Humoral Hypercalcemia of Malignancy
This article is included in the Oncology gateway.
This article is included in the Manipal Academy of Higher Education gateway.
Neuroendocrine Tumor, Duodenal NET, Humoral Hypercalcemia of Malignancy, Parathyroid Hormone Related Protein, Paraneoplastic Syndromes, Humoral Hypercalcemia of Malignancy
This revised version substantially expands and refines the clinical narrative and discussion compared with the previously published version. Additional longitudinal details of disease progression, imaging findings, and therapeutic sequencing—including the rationale for surgery, somatostatin analogues, everolimus, and PRRT—have been incorporated to improve clarity and reproducibility. The diagnostic workup and management of severe hypercalcemia are described in greater depth, with clearer attribution to PTHrP-mediated humoral hypercalcemia of malignancy and exclusion of alternative causes. The discussion has been significantly strengthened by integrating recent evidence, including updated guideline perspectives and data from contemporary trials, to better contextualize treatment decisions. Overall, this version provides a more comprehensive clinical, pathophysiological, and therapeutic analysis, enhancing the educational and scientific value of the report.
See the authors' detailed response to the review by Ahmed Saad Abdlkadir
Neuroendocrine tumors (NETs) denote a heterogeneous group of neoplasms derived from cells of predominant neuroendocrine differentiation. Although they can arise from various sites throughout the body, duodenal NETs account for only 2-3% of all gastrointestinal NETs and approximately 1-2% of all duodenal tumors.1,2 These tumors are generally characterized by their slow growth and indolent behavior; however, some may exhibit aggressive features with metastatic potential.3
Humoral hypercalcemia of malignancy (HHM) is a paraneoplastic syndrome most commonly associated with solid organ malignancies like squamous cell carcinomas of the lung and head & neck, along with breast, ovarian, renal, and bladder carcinomas. Also, hematological malignancies like Non-Hodgkins Lymphoma (NHL) frequently report paraneoplastic hypercalcemia.4 Various mechanisms by which HHM can occur are by:
• Secretion of Parathyroid Hormone related peptide (PTHrp) by the tumor,
• Osteolytic metastases and,
• Production of 1,25-dihydroxyvitamin D (calcitriol) by the tumor.
Parathyroid hormone-related peptide (PTHrP) mimics the physiological effects of parathyroid hormone, leads to increased bone resorption and calcium reabsorption in the kidney.5 The association between NETs, particularly those of duodenal origin, and HHM is exceedingly rare, with only a handful of cases reported in the literature.6,7
This case report describes a patient with a metastatic duodenal neuroendocrine tumor presenting with humoral hypercalcemia of malignancy, highlighting the clinical challenges in diagnosis and management of this unusual presentation. This case underscores the importance of considering NET as a potential etiology in patients with unexplained hypercalcemia.8 Additionally, we discuss the unique pathophysiological mechanisms involved in NET-induced HHM and review the current therapeutic approaches for both the underlying malignancy and its metabolic complications.9,10
A 70-year-old Indian male, retired businessman by occupation, with a history of hypertension and diabetes mellitus, was diagnosed to have Duodenal Neuroendocrine Tumor in 2018.
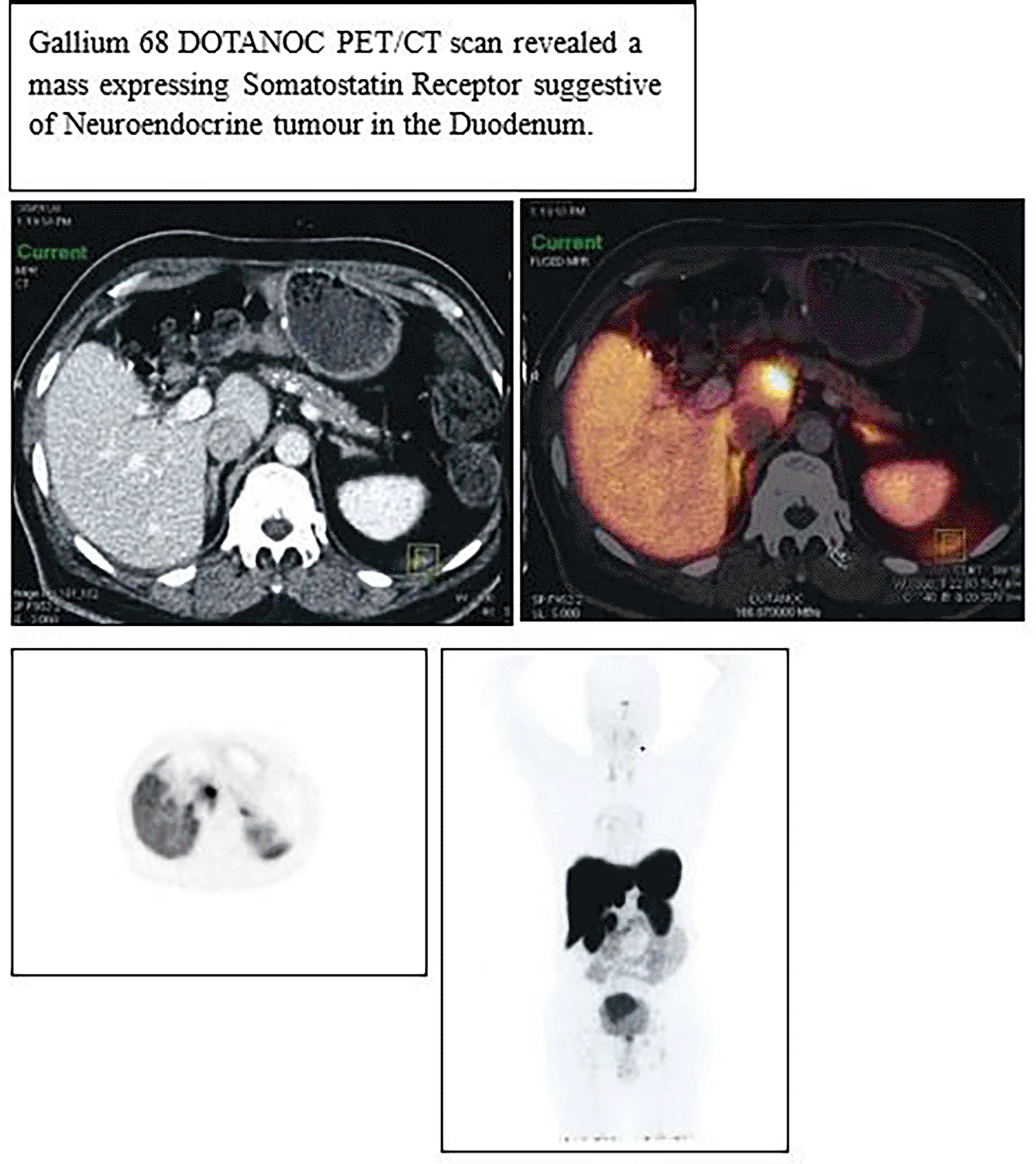
At initial staging in March 2018, the patient was found to have Duodenal NET with regional lymph node involvement but without any evidence of metastasis, TNM Stage T2N1M0 (according to AJCC 8th Edition). It was a well-differentiated neuroendocrine tumor of the duodenum ( Figure 1), grade 2, with a Ki67 index of 18.2% and mitotic rate of 12 mitosis per 10 high-power fields. For this condition, he underwent distal gastrectomy with D1 duodenal resection in May 2018, followed by medical management with Octreotide (Long-Acting Release) LAR injection 30 mg once a month intramuscularly.
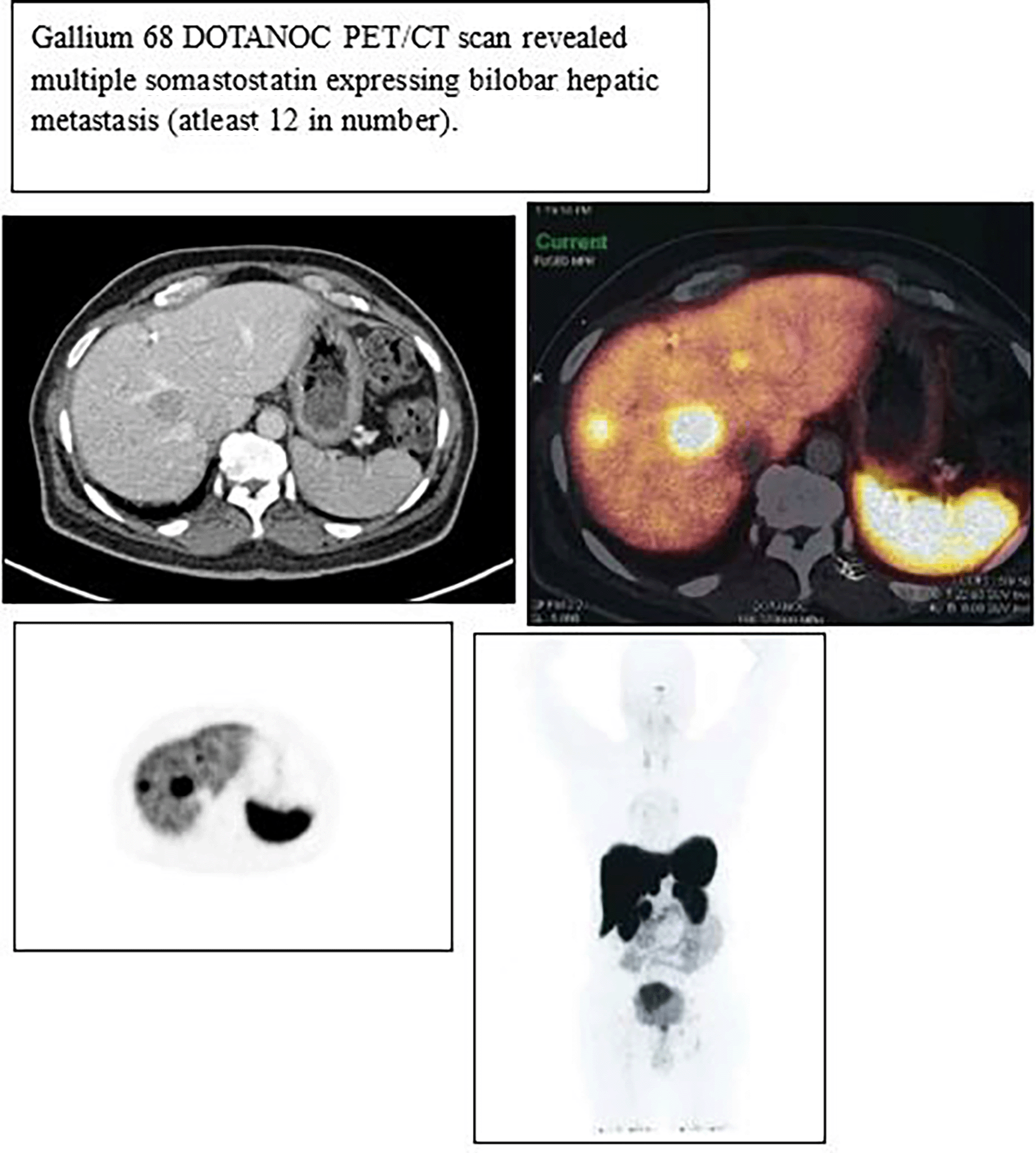
Despite initial interventions, subsequent Gallium 68 DOTANOC PET/CT scan in March 2020 revealed disease progression, demonstrating multiple somatostatin-expressing arterially enhancing metastases (approximately 12) in bilateral lobes of the liver and a peripancreatic node (as shown in Figure 2). For this he was managed with monthly Octreotide LAR injections and Tablet Everolimus 5 mg once daily at night.
Due to the increase in the somatostatin expression and the size of the liver lesions as well as peripancreatic node, he was initiated on Peptide Receptor Radionuclide Therapy (PRRT) in August 2021. Octreotide and Everolimus were withheld.
He received 200 mCi (or 7.4 GBq) of Lutetium 177 DOTATATE in each cycle of PRRT Therapy. A total of 4 such cycles were given with an interval of 8 weeks between two consecutive cycles. The treatment was completed by March 2022. He tolerated the therapy well without any adverse events. Complete blood count, Renal function tests and Liver function tests were regularly monitored and were well within normal limits.
Follow up Gallium 68 DOTANOC PET/CT scan in April 2022 revealed decrease in the somatostatin expression and the size of the liver lesions as well as peripancreatic node. He was maintained on Octreotide LAR injections and Tablet Everolimus.
However, in October 2022, patient presented to the emergency room with a two-day history of unresponsiveness. Physical examination revealed gasping respirations, a Glasgow Coma Scale (GCS) score of 3/15, severe hypoxia with an oxygen saturation of 74% on room air, and signs of severe dehydration.
Initial laboratory investigations as depicted in Table 1 were significant for anaemia, thrombocytopenia, severe hypercalcemia with serum calcium levels of 13.1 mg/dL (corrected calcium 14.1 mg/dL), and acute kidney injury. Corrected Calcium was calculated as per the following formula20:
Corrected Calcium (mg/dL) = 0.8 × (Normal albumin (g/dL) - patient’s albumin(g/dL)) + serum calcium (mg/dL)
Immediate management included endotracheal intubation for airway protection and aggressive intravenous fluid resuscitation.
Given the striking hypercalcemia, further workup was undertaken to determine its etiology (as elaborated in Table 2).
| Phosphate | 3.4 mg/dL (normal) |
| Vitamin D total | 8.47 ng/ml (low) |
| iPTH (Intact Parathyroid Hormone) | 6.62 pg/ml (low) |
| PTHrp (Parathyroid Hormone-Related Protein) | 252 pmol/L (high) |
Multiple myeloma, a common cause of hypercalcemia, was ruled out through serum protein electrophoresis (absence of M Band) and free light chain assay showing normal Kappa/Lambda ratio of 1.2.
No osteolytic bony metastasis were noted in the PET/CT scans and ALP levels being normal ruled out tumor induced osteolysis as the cause for hypercalcemia.
Low levels of vitamin D and PTH ruled out Ectopic calcitriol or PTH secretion by the tumor.
The definitive clue to the etiology of hypercalcemia came with markedly elevated parathyroid hormone-related protein (PTHrP) levels measured at 252 pmol/L, confirming the diagnosis of humoral hypercalcemia of malignancy (HHM) secondary to the metastatic duodenal NET.
Also, the patient’s persistent altered sensorium prompted an MRI brain which showed chronic lacunar infarcts in the right frontal periventricular white matter, small vessel ischemic changes in bilateral frontoparietal, periventricular, subcortical, and deep white matter and age-related cerebral atrophy.
EEG (Electroencephalogram) revealed mild to moderate diffuse electrophysiological dysfunction with intermixed beta activity suggestive of hypoxic-ischemic encephalopathy.
In order to address the severe hypercalcemia, subcutaneous Calcitonin 250 IU BD was initiated, and intravenous Ibandronate therapy was also given. Intravenous hydration continued at 1.5-2 litres per day. Response to therapy was monitored by serially checking the calcium levels as shown in Table 3.
| Day 3 | Day 4 | Day 5 | |
|---|---|---|---|
| Urea | 91 mg/dL | 96 mg/dL | 98 mg/dL |
| Creatinine | 1.98 mg/dL | 2.01 mg/dL | 1.99 mg/dL |
| Calcium | 14 mg/dL | 13.8 mg/dL | 14.1 mg/dL |
| Corrected Calcium | 15 mg/dL | 14.8 mg/dL | 15.1 mg/dL |
However, hypercalcemia persisted, so calcitonin was increased to 250 IU intravenously TID and intravenous Dexamethasone 4 mg TID was added. Ultimately due to the refractory nature of hypercalcemia and the presence of acute kidney injury, daily hemodialysis was initiated. Following this calcium levels started to decline as shown in Table 4.
| Day 6 | Day 7 | Day 8 | |
|---|---|---|---|
| Urea | 92 mg/dL | ||
| Creatinine | 2.04 mg/dL | ||
| Calcium | 12.2 mg/dL | 11.4 mg/dL | 11.8 mg/dL |
| Corrected Calcium | 13.2 mg/dL | 12.4 mg/dL | 12.8 mg/dL |
Neuroprotective measures, including Piracetam administration, were instituted for the hypoxic-ischemic brain injury.
Despite comprehensive multidisciplinary interventions, the patient’s neurological status failed to improve. The irreversible hypoxic brain damage ultimately led to the patient’s demise.
This case highlights the rare occurrence of humoral hypercalcemia of malignancy (HHM) in a patient with metastatic duodenal neuroendocrine tumor (NET). While hypercalcemia is a common paraneoplastic manifestation in various malignancies, affecting approximately 20-30% of cancer patients, its association with NETs is notably uncommon, with an estimated prevalence of only 1-2%.4,6 Among the spectrum of NETs, pancreatic NETs have been more frequently implicated in HHM compared to those of gastrointestinal origin, making our patient’s presentation particularly noteworthy.8
The pathophysiology of HHM in our patient was attributed to elevated Parathyroid Hormone- related Protein (PTHrP) levels, which is consistent with published literature. PTHrP shares significant homology with parathyroid hormone (PTH) at the N-terminal region, allowing it to bind and activate the PTH receptor, thereby inducing hypercalcemia through increased osteoclastic bone resorption and enhanced renal tubular calcium reabsorption.11 Milanesi et al. reported six cases of PTHrP-secreting NETs causing HHM, predominantly originating from the pancreas, with only one case arising from the duodenum.10 The authors observed PTHrP levels ranging from 21 to 169 pmol/L, which is notably lower than the 252 pmol/L documented in our patient, suggesting particularly aggressive PTHrP secretion.
The clinical presentation of our patient with severe altered consciousness and profound hypercalcemia (corrected calcium of 14.06 mg/dL) aligns with the findings of Goldner et al., who demonstrated that neurological manifestations predominate when serum calcium exceeds 14 mg/dL.12 Similarly, Kamp et al. in their cohort analysis of GEP-NETs (Gastroenteropancreatic - NETs), found that severe hypercalcemia (>14 mg/dL) was associated with a significantly poorer prognosis and rapid clinical deterioration, consistent with our patient’s course.13
Interestingly, HHM in NETs appears to correlate with higher tumor grade and proliferative indices. Our patient’s tumor exhibited a Ki-67 index of 18.2%, classifying it as grade 2. This observation is consistent with the findings of Ilias et al., who reported a positive correlation between tumor grade and incidence of paraneoplastic syndromes, including HHM, in a cohort of 90 patients with NETs.9 This relationship may be attributed to the dedifferentiation process, where higher-grade NETs lose their typical neuroendocrine characteristics and acquire the ability to produce atypical hormones, including PTHrP.
Abdlkadir et al. also reported a well-differentiated gastroenteropancreatic neuroendocrine tumor exhibiting aggressive behavior and progression despite standard therapies, underscoring the marked biological heterogeneity of NETs. Similar to our case, disease advancement occurred despite strong somatostatin receptor expression, ultimately warranting escalation to PRRT. However, the authors described a Grade 1 ileal NET with rare leptomeningeal and extraocular muscle metastases at presentation, whereas our patient had a Grade 2 duodenal NET initially treated with curative-intent surgery and later developed hepatic and nodal metastases. Furthermore, our case was complicated by humoral hypercalcemia of malignancy, a rare paraneoplastic feature. Collectively, these observations highlight that tumor grade alone does not reliably predict clinical aggressiveness and support timely consideration of PRRT in progressive, receptor-positive NETs.14
The management approach for our patient involved a multifaceted strategy targeting both the hypercalcemia and the underlying malignancy. However, despite aggressive interventions including bisphosphonate therapy, hemodialysis, and previous PRRT, the patient’s outcome remained poor. This therapeutic challenge is echoed in the literature, with Ralston et al. demonstrating that survival in patients with NET-associated HHM is significantly shorter compared to those with HHM from other malignancies.15 The authors attributed this to the rapid progression of metastatic disease and the refractory nature of hypercalcemia in these cases.
The role of Peptide Receptor Radionuclide Therapy (PRRT) in the management of metastatic NETs with paraneoplastic manifestations warrants further discussion. Kwekkeboom et al., in their analysis of 504 patients with GEP-NETs (Gastroenteropancreatic NETs) treated with PRRT, noted resolution of paraneoplastic syndromes in 39% of cases, suggesting potential efficacy.16 However, our patient had already received four cycles of PRRT before developing HHM, indicating possible treatment resistance or disease evolution. This observation aligns with the findings of Strosberg et al., who documented that approximately 30% of initially responsive NETs develop resistance to PRRT over time, particularly those with higher proliferative indices.17
Although early PRRT has recently shown significant benefit in newly diagnosed, advanced grade 2–3 well-differentiated GEP-NETs, as demonstrated in the NETTER-2 phase III trial (Singh et al., 2024), these data were unavailable when our patient was managed in 2018. At that time, international guidelines advocated a stepwise approach, emphasizing surgery for localized disease followed by somatostatin analogues and targeted therapies, with PRRT reserved for later lines. Accordingly, our patient received guideline-concordant management, with PRRT introduced after disease progression. This case highlights the evolving therapeutic paradigm and supports earlier consideration of PRRT in biologically aggressive NETs.18
From a molecular perspective, the mechanisms driving PTHrP secretion in NETs remain incompletely understood. Wysolmerski et al. postulated that the transcription factor RUNX2 plays a crucial role in regulating PTHrP expression in neuroendocrine cells.19 Additionally, genomic analyses by Javle et al. identified alterations in the Calcium-Sensing Receptor (CaSR) gene in a subset of PTHrP-secreting tumors, potentially contributing to dysregulated calcium homeostasis. These molecular insights may offer potential therapeutic targets for future management strategies.
It is noteworthy that our patient exhibited low vitamin D levels concurrent with hypercalcemia, a finding reported by Galitzer et al. in approximately 40% of patients with HHM.20 This vitamin D deficiency may represent a compensatory mechanism to mitigate hypercalcemia or could reflect nutritional compromise in advanced malignancy. Additionally, the acute kidney injury observed likely resulted from the combined effects of hypercalcemia-induced renal vasoconstriction, volume depletion, and potential nephrotoxicity from prior treatments, as detailed by Sternlicht and Glezerman in their review of renal complications in patients with malignancy.21
Our case adds to the limited literature on HHM in duodenal NETs and emphasizes the importance of considering this rare paraneoplastic manifestation in the differential diagnosis of hypercalcemia in patients with NETs. The exceptionally high PTHrP level (252 pmol/L), the grade 2 histology, and the refractory nature of both the hypercalcemia and the underlying malignancy to established therapies, including PRRT, represent distinctive features of our case.
Despite multimodal interventions including bisphosphonate therapy, hemodialysis, and prior PRRT, the patient’s outcome was poor, reflecting the challenges in managing this paraneoplastic manifestation. This case underscores the importance of considering PTHrP- mediated hypercalcemia in the differential diagnosis of hypercalcemia in NET patients, even when other more common etiologies such as bony metastases are typically encountered. Early recognition, prompt intervention, and continued research into molecular mechanisms underlying PTHrP secretion in NETs are essential to improve outcomes in this rare but clinically significant entity.
Moving forward, we recommend routine screening for hypercalcemia in all patients with metastatic NETs and consideration of PTHrP levels in those with unexplained hypercalcemia, even in the absence of bone metastases.
CARE Guidelines have been used for the reporting of our case report. It is available as a supplementary document in the external repository and the link for the same has been included in the References section.22
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)