Keywords
Genetics, compound heterozygous, genome analysis, trio, phasing, reproducibility
This article is included in the Software and Hardware Engineering gateway.
This article is included in the Bioinformatics gateway.
Genetics, compound heterozygous, genome analysis, trio, phasing, reproducibility
We updated the abstract to get to the point more quickly, to mention the tools used for each major step, and to clarify that MAF and CADD are used as part of variant filtering. We updated the manuscript to mention that whole-exome and targeted exome data can also be used. We changed the way we filter the variants to be more simple but also more strict. Now, CADD and MAF scores must be available for all variants, variants must be in exonic regions, and variants can have either “MED” or “HIGH” putative impact. This update to filtering did not alter the results in our example analyses. There was an error in “identify_homAlt_variants.py” that required a gene to have 2 variants in order to be written to the output file. This has been fixed, and it slightly changed the example results. Originally we had reported only 1 gene with a homozygous alternate variant in the example dataset. However, after updating the script and running it again, we identified 2 genes that have a homozygous alternate variant. The manuscript has been updated accordingly. In response to reviewer comments, we have updated the “gemini_load.py” script with an optional argument to allow users to import a file that was annotated with VEP. Since SnpEff is used as part of the pipeline, the script defaults to snpEff, but if a user chooses to use VEP outside of the pipeline, then they can still use the remaining steps of the pipeline. We have updated Figure 1 to show step numbers. We have updated the manuscript to say “global minor allele frequency” when first referenced. Clarifications about phasing were made to both the main text and example PDF. Other minor updates were made to the example PDF and manuscript for added clarity and explanation.
See the authors' detailed response to the review by Soohyun Lee
See the authors' detailed response to the review by Sushant Patil
A compound heterozygous (CH) variant occurs when a person inherits two alleles, one from each parent, and these alleles are located at different positions within the same gene1. The compound effects of these alternate alleles may lead to phenotypic effects as seen in some cases of human disease, including ataxia telangiectasia, NGLY1 deficiency, and various types of pediatric cancer2–4. For example, CH variants in the mismatch repair gene, MSH6, have been identified in pediatric patients with colorectal cancer, medulloblastoma, high-grade glioma, glioblastoma, non-Hodgkin’s lymphoma, and acute lymphoblastic leukemia4. To detect CH variants in next-generation sequencing data, it is necessary to differentiate between paternally and maternally derived nucleotides1. Laboratory-based methods such as fosmid-pool-based or linked-read sequencing can be used; however, if DNA libraries are prepared and sequenced without regard to nucleotide inheritance (as is done in most sequencing projects), computational methods can help determine parental inheritance through haplotype estimation (“phasing”)5–7.
Available phasing tools require specific input file types (such as VCF or Plink files) and reference files which are not standardized across different phasing software. In addition, many phasing programs require that input files have been aligned to a specific reference genome, do not contain multiallelic positions, are free of repeat positions, and that each chromosome be phased separately8–10. Figuring out how to prepare files for phasing can be challenging as passing files from program to program may result in unforeseen incompatibilities. Additionally, installing some programs can be challenging because of operating-system specific installation processes and software dependencies.
We have designed Compound Heterozygous Variant Identification Pipeline (CompoundHetVIP) to help researchers overcome these time-consuming challenges when identifying CH variants. CompoundHetVIP encapsulates specific versions of existing tools, required software dependencies, and custom Python scripts into a cohesive computational environment packaged as a Docker image11. Accordingly, researchers need only install the Docker software and download the CompoundHetVIP Docker image to begin performing CH, homozygous alternate, and de novo analyses at the command line. Furthermore, because the source code for CompoundHetVIP is publicly available, other researchers will be able to reproduce the analyses and investigate the specific methodologies used.
The functionality of CompoundHetVIP is divided into a series of 13 steps (Figure 1). For each step, a Python script is executed within a Docker container. These scripts provide logic for processing data files and invoking third-party tools. By breaking the pipeline into 13 steps, users have flexibility to perform the steps that are most relevant to their analysis. For example, researchers can use input data for an affected individual only or for a trio (an affected individual and both parents). If parental data are unavailable and the variant positions within the VCF file correspond to genome build GRCh37, users may skip the first three steps. A detailed, step-by-step guide is available on GitHub and as Extended data12.
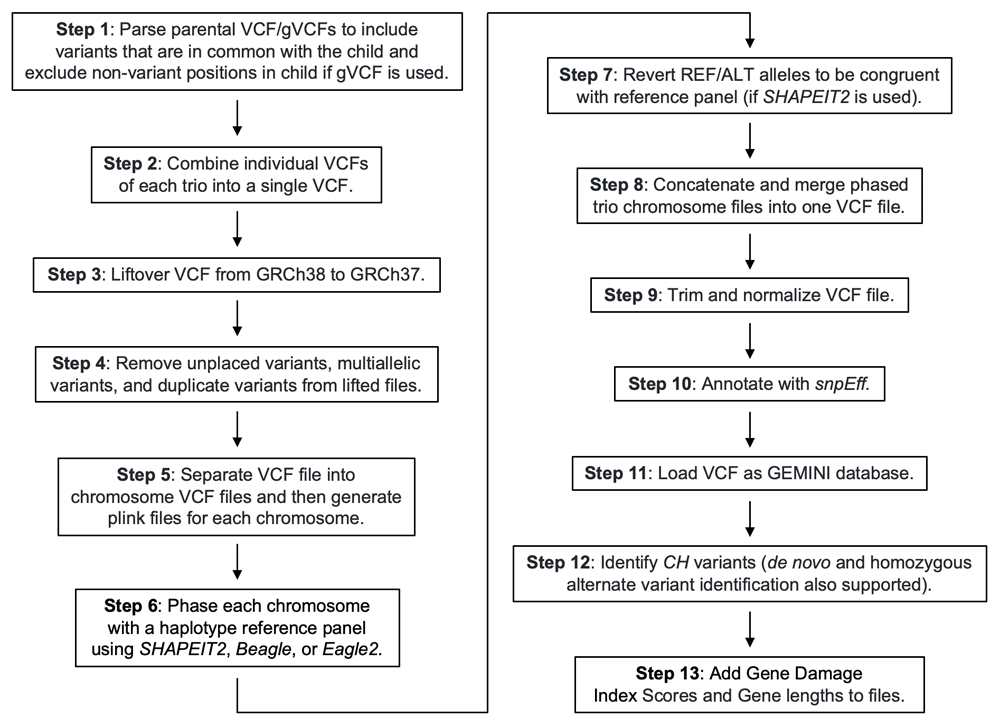
For step 1, the inputs can be either Variant Call Format (VCF)13 or gVCF14 files that were generated from whole-genome, whole-exome, or targeted exome sequencing data. VCF files contain variant sites only, whereas gVCF files include non-variant sites, too. For each parent of a trio being evaluated, our script retains nucleotide positions that are in common with the child. When gVCF files are used (whether for trios or individuals), our script removes all non-variant sites for the child (but retains these for the parents to support determination of CH status). When applied to trio data, some phasing tools, such as SHAPEIT28, require a single input file for each trio. Therefore, in step 2 (used only when working with trios), we combine the variant files for each member of a trio into a single VCF file using either BCFtools (VCF input files)15 or GATK4 (gVCF input files)14. If GATK4 is used, joint-genotyping is also performed on the trio VCF.
The remaining steps can be applied either to trios or individuals. Some phasing and annotation programs require that data be aligned to genome build GRCh37; thus, we use this reference genome as our standard. For variant files that have been aligned to genome build GRCh38, step 3 uses Picard Tools16 to convert the data to GRCh37 positions using a lift-over process. During lift-over, some sites may be present in GRCh38, but their exact position in GRCh37 is unknown. To avoid ambiguity, these sites are removed during step 4. This step also removes positions that are multiallelic or duplicated to maintain compatibility with programs such as Plink217,18 and SHAPEIT2 (used in steps 5 and 6, respectively). For trio VCF files, sites that contain missing genotype information (i.e. “./.”) for both parents are removed to improve phasing accuracy.
CompoundHetVIP can perform phasing using SHAPEIT2, Eagle210, and/or Beagle9. Each of these programs requires that each chromosome be phased independently. Additionally, when using SHAPEIT2, it is recommended that PLINK files (.bed, .bim, .fam) be used as input for phasing. Therefore, step 5 divides a VCF file by chromosome into multiple files and creates the necessary PLINK files for each chromosome (when SHAPEIT2 is used for phasing).
Step 6 phases the variants in each chromosome using default parameters for the phasing program chosen by the user. We recommend using SHAPEIT2 because it can applied either to trios or individuals. When parents’ genotypes are available, this program uses Mendelian logic for phasing and a population-based haplotyped reference panel when the phase of the child cannot be determined from Mendelian logic alone (i.e. both parents and child are heterozygous). In addition, if a parent is missing genotype information at a position, SHAPEIT2 imputes the missing information. All supported phasing programs integrate the 1000 Genomes Project phase 3 haplotype reference panel19 and do not require sequence alignment files (.bam), such as those required by read-based programs20,21. In some scenarios, SHAPEIT2 switches the REF and ALT alleles. Therefore, step 7 ensures that the REF/ALT alleles of the phased VCF files are congruent with those of the reference genome. Also, sites with Mendelian errors are removed.
To make subsequent analysis of the phased files easier, step 8 concatenates all phased chromosomes into a single file. If a user is analyzing multiple trios (or individuals), this script can also merge the data for these trios (or individuals) into a single VCF file.
Step 9 normalizes VCF files as recommended by GEMINI22 (used in step 11). Normalization involves left-alignment and trimming of variants23. This process helps ensure that variants are represented at their left-most position, with as few nucleotides as possible, and unambiguously. This step uses vt tools23. In step 10, SnpEf24 provides information about the effects of variants on function for known genes. Then, in step 11, GEMINI22 loads the annotated VCF into a relational database (GEMINI can also load files annotated with Variant Effect Predictor (VEP)25, although VEP is not available as part of our pipeline). Step 12 uses a custom Python script to extract CH variant data from the database. Our provided script identifies CH variants and filters the data based on user-set thresholds for global minor allele frequency (MAF) and Combined Annotation Dependent Depletion (CADD) scores26. Variants with a MAF less than or equal to the user-set threshold, CADD score greater than or equal to the user-set threshold, exonic classification, and “HIGH” or “MED” putative impact severity are included in the final output. We consider the variants in the final output as candidates for further evaluation. For step 12, we provide two additional scripts that identify homozygous alternate variants and de novo variants using the same user-set thresholds as those described above.
Finally, in step 13, we add Gene Damage Index (GDI) scores27 and gene-length information to the output files. GDI scores quantify accumulated mutational damage in healthy populations as a way to predict whether genes are likely to have disease-causing variants. Genes of longer length (e.g. TTN, MUC5B) tend to have more total damage but typically less disease-causing damage than shorter genes.
Because CompoundHetVIP executes all scripts within a Docker container, it can be executed on all major operating systems that are commonly used for scientific computing. Depending on input file sizes, the hardware needed to execute CompoundHetVIP will vary from user to user. Certain tasks, such as phasing (step 6), can be memory intensive. A minimum of 40 GB memory is recommended. When creating a relational database with GEMINI (step 11), there is no minimum processing core recommendation, but multiprocessing can significantly speed up the time it takes to load the database. Users can specify how many processing cores GEMINI can use when executing step 11.
We applied CompoundHetVIP to high-confidence, VCF data that were generated with whole-genome sequencing data from an Ashkenazim trio available through the Genome in a Bottle Consortium28. During step 6, we used SHAPEIT2 to phase the data. In the child of this trio, we identified a CH variant in two genes (FLNB and TTN) using a MAF threshold of 0.01 and a CADD score threshold of 15. Genes with a GDI score less than or equal to 13.84 are classified as being more likely to have disease-causing damage from variants27. FLNB (6.2) was lower than this threshold but TTN (42.9) was not. FLNB has an important role in cytoskeleton development and variations in this gene have been associated with many skeletal disorders29,30.
In addition, we identified two homozygous alternate variants: one in TBC1D2 and the other in TOX2, using the same MAF and CADD thresholds that we used for CH variant identification. TBC1D2 and TOX2 had GDI scores of 9.7 and 4.4, respectively. TBC1D2 codes for a GTPase-activating protein and is involved in E-cadherin degradation31. The role of this gene and how it may relate to human disease is not yet fully understood. TOX2 is a transcription factor that helps drive the development of T follicular helper (Tfh) cells32. Tfh cells are an important part of humoral immunity.
Using the same MAF and CADD thresholds described above, we sought to identify de novo variants in this trio. However, none passed these thresholds.
CompoundHetVIP provides the necessary tools for CH variant identification using VCF or gVCF files as initial input and is executed within a Docker container, which allows for cross-platform compatibility and reproducibility. CompoundHetVIP involves 13 steps (Figure 1) that include a breadth of tasks such as file aggregation, VCF liftover, joint-genotyping, file conversion, phasing, variant normalization, annotating, and variant identification. Our results highlight that potentially damaging CH and homozygous alternate variants are observed in seemingly healthy individuals. However, we anticipate that most researchers will use CompoundHetVIP to identify variants in individuals with a known disease.
VCF data used to generate the results were from an Ashkenazim trio, freely-available through the Genome in a Bottle Consortium at ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/AshkenazimTrio/27:
Zenodo: dmiller903/CompoundHetVIP: CompoundHetVIP - v1.1. https://doi.org/10.5281/zenodo.447768612.
This project contains the following extended data:
Software available from: https://hub.docker.com/r/dmill903/compound-het-vip
Source code available from: https://github.com/dmiller903/CompoundHetVIP
Archived source code at time of publication: https://doi.org/10.5281/zenodo.447768612.
License: MIT
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)