Keywords
Invasive ductal carcinoma, prognostic biomarkers, hub genes, microarray technology, differentially expressed genes, GEO, GEO2R
This article is included in the Oncology gateway.
This article is included in the Bioinformatics gateway.
This article is included in the Bioinformatics in Cancer Research collection.
Invasive ductal carcinoma, prognostic biomarkers, hub genes, microarray technology, differentially expressed genes, GEO, GEO2R
Breast cancer (BC) is the most prevalent diagnosed neoplasm in women worldwide and one of the most important causes of death among them.1,2 According to The Global Cancer Observatory, in 2020, there were more than 2.3 million new cases worldwide. On the other hand, BC deaths are reported more frequently in countries such as Melanesia, Western Africa, Micronesia/Polynesia, and the Caribbean.3
BC has been categorized into two major histological types, invasive ductal carcinoma (IDC) and invasive lobular carcinoma (ILC),4 with IDC being the most common (80%)5; this neoplasm begins in the cells lining a milk duct in the breast, from there, the cancer cells invade the wall of the duct, and grows into nearby breast tissues. At this point, it may have the ability to spread (metastasize) to other parts of the body through the lymphatic system and bloodstream.6
The clinicopathological characteristics and differences between IDC and ILC and BC prognosis have been well described.6 Identified prognostic factors of BC have been crucial in the diagnostic markers, workup, and treatment of this pathology7; these include the hormone receptors [estrogen receptor (ER)/progesterone receptor (PR) positive], human epidermal growth factor receptor 2 (HER2/neu), and germline mutations in BReast CAncer gene 1 (BRCA1) or BReast CAncer gene 2 (BRCA2), which are associated with an increased risk of BC incidence.5 Prognostic markers are also helpful in determining the effectiveness of the established intervention (surgical or pharmacological treatment), the probability of recurrence, and the establishment of additional follow-up and treatment strategies.8
Despite efforts to identify biomarkers for BC, due to its heterogeneity and its high capacity for metastasis, an increasing percentage of patients are demanding personalized treatments,9 which makes it necessary for the discovery of novel biomarkers for diagnosis and prognosis that allow for an early evaluation of the development of the pathology to formulate effective diagnosis and treatment strategies.10–12
Nowadays, the analysis of gene expression profiles [verification of differentially expressed genes (DEGs)] using bioinformatics tools has represented a notable advance in research in clinical oncology aimed at the identification of genes related to tumors, new molecular markers of diagnosis and prognosis, and evaluation of therapeutic effects among others.13 DEGs and protein-protein interaction network (PPI-net) analysis have been widely used to identify biomarkers and potential drug targets. Open access databases such as Gene Expression Omnibus (GEO) are broadly employed as microarray resources for this purpose.14
Previous studies have identified prognostic genes from ductal carcinoma in situ (DCIS), such as Fibroblast growth factor 2 (FGF2), Growth arrest-specific protein 1 (GAS1), and Secreted frizzled-related protein 1 (SFRP1) using GEO15; however, currently, IDC is little understood from the genomic point of view, and there are no studies from the analysis of expression of genes using bioinformatic methods. Thus, this study aimed to identify new prognostic genes of this type of BC using an integrated bioinformatics approach.
The two expression profiling datasets from BC, GSE29044 and GSE632291, were downloaded from GEO (RRID:SCR_005012), which were based on the platforms [GPL570 (HG-U133_Plus_2 - Affymetrix Human Genome U133 Plus 2.0 Array)] and [GPL4091 (Agilent-014693 Human Genome CGH Microarray 244A (Feature number version)], respectively. GSE29044 was a collection that analyzed the whole-genome mRNA expression profile from 73 patients with tumors and 36 adjacent disease-free tissues using the Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays.16 On the other hand, GSE32291 was analyzed using whole-genome CGH arrays from Agilent 394 invasive ductal breast carcinomas and 20 normal breast biopsies.
The inclusion criteria used for the selection of the samples were that they were both ER and PR positive and a wild type of strain. The MeSH (RRID:SCR_004750) terms used for the selection of the datasets were (“carcinoma, ductal, breast” [MeSH Terms] OR invasive ductal carcinoma [All Fields]) AND “Homo sapiens”[porgn]).
To verify the intra-group data repeatability per each group of datasets, as proposed by Xu et al. (2019), we developed a Pearson’s correlation test using the R programming language, R Project for Statistical Computing (RRID:SCR_001905). The degree of correlations between all samples from the same dataset was visualized by heat maps built in R.17
DEGs in the first three stages of the IDC were obtained by online analysis in GEO2R (RRID:SCR_016569), which is an interactive online tool from GEO that finds DEGs through the comparison of the original submitter-supplied processed data tables using the GEOquery (RRID:SCR_000146) and limma R packages from the Bioconductor project.18–20 Initially, the experimental groups were built from the datasets, grouping the samples as tissues with IDC and controls (adjacent disease-free tissues). A comparative analysis was carried out for each IDC stage evaluated.1–3 The cut-off criterion was P < 0.05 and a fold-change among ≥ 1.5 or ≤ 1.5. Volcano plots with the DEGs found were drawn in GEO2R. An intersection analysis between DEGs extracted from the three stages assayed was made by drawing Venn diagrams, delineated in the functional enrichment analysis tool (FunRich).17,21
A PPI-net was built with the DEGs product of the intersection analysis between the three IDC stages evaluated, using the online Search Tool for the Retrieval of Interacting Genes [STRING (RRID:SCR_005223)], thus identifying the prognostic candidate genes (hub genes). Next, through the software Cytoscape (RRID:SCR_003032) (version 3.8.0),22 the PPI-net was visualized; on the other hand, the MCODE App (RRID:SCR_015828) (Molecular complex detection tool; version 1.6.1) was used to identify the most important module of the network.23 The criteria for MCODE analysis were a degree of cut-off of 2, scores >5, a maximum depth equal to 100, a node score cut-off of 0.2, and a k-score of 2. Genes with degrees ≥10 were selected as hub genes.17,19
After the identification of the main module of the network, the top 10 central genes were evaluated through the cytoHubba (RRID:SCR_017677) application of Cytoscape,24 using the five most reported calculation methods: Degree, EcCentricity, closeness, Maximum Neighborhood Component (MNC), and Maximal Clique Centrality (MCC).25 An intersection analysis was performed between the hub genes identified for each method in a virtual tool VennDiagram image GP.26 Finally, a functional enrichment analysis of the hub genes identified in FunRich was performed, and through the Kyoto Encyclopedia of Genes and Genomes (KEGG) (RRID:SCR_012773) we analyzed the enrichment around the molecular function.27,28
We assayed the expression patterns of hub genes between different stages of IDC based on Gene Expression Profiling Interactive Analysis (GEPIA) (RRID:SCR_018294), a web server for cancer and normal gene expression profiling and interactive analyses.29
All analyses were conducted in GraphPad Prism (RRID:SCR_002798) (version 8.0.2) [free alternative, JASP (RRID:SCR_015823) (version 16.3)] and RStudio (RRID:SCR_000432). One-way analysis of variance (ANOVA) was used for comparing the mean between groups in the analyses conducted in GEPIA. P<0.05 was considered to indicate a statistically significant difference.
R script for GSE29044 and GSE632291 can be found as Underlying data.62–67 Pearson’s correlation coefficient showed that both datasets (GSE29044 and GSE32291) had a strong correlation among the samples from the control group and IDC (Supplementary Figure S1, which can be found as Extended data70). Next, we classified the samples of the datasets per stage,1–3 and, through GEO2R, a volcano plot analysis was performed to identify the DEGs in the three stages assayed. Nodes that conformed to the cut-off criterion (fold-change ≥1.5 or ≤-1.5, and a P<0.05) were represented in blue or red color; the first represented downregulated DEGs and the red the upregulated DEGs in IDC samples, regarding the controls (Figure 1a).68 An intersection analysis in FunRich was made with the DEGs from each dataset per stage, and those genes were used to find the common DEGs in the three stages assayed; we found 1,085, 3,213, and 3,477 common DEGs in stages 1, 2, and 3, respectively (Supplementary Figure S2, which can be found as Extended data71). We also found 724 common DEGs in the three stages (Figure 1b) (P<0.05).
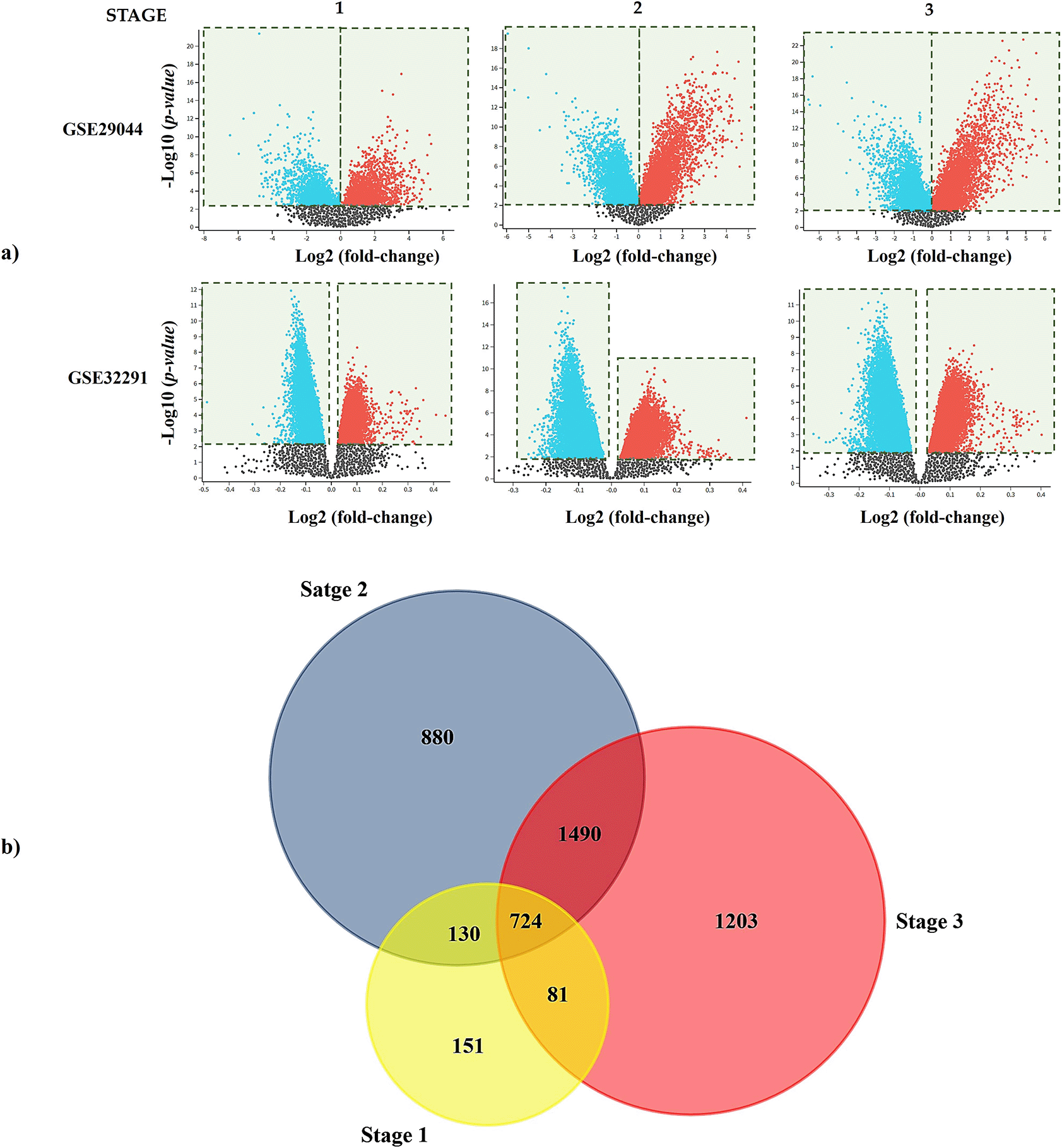
a) Volcano plots obtained in GEO2R show the difference in gene expression between tissues of IDC and controls. The X and Y-axis represent the fold-change and the P-value (log-scaled). Each symbol represents a different gene; red and blue symbols represent upregulated and downregulated genes. b) Venn diagram showing the shared genes per stages assayed. DEG, differentially expressed gene; IDC, invasive ductal carcinoma.
From 724 common DEGs, a PPI network was built in STRING using the following parameters: medium confidence of > 0.4 (minimum required interaction score) and that the network will only display the query proteins; Supplementary Figure S3, which can be found as Extended data,72 described the network features. Next, we identified the most significant PPI-net module by the MCODE app from Cytoscape, which had 73 edges, 17 nodes, and a score of 9.125 (Figure 2a); from it and using the five most reported calculation methods of cytoHubba (Degree, EcCentricity, MCC, MNC, and Closeness) we identified seven hub genes by intersection analysis [WW domain-containing E3 ubiquitin-protein ligase 1 (WWP1), STIP1 homology and U-box containing protein 1 (STUB1), F-box and WD repeat domain containing 7 (FBXW7), kelch like family member 13 (KLHL13), ubiquitin-conjugating enzyme E2 Q1 (UBE2Q1), tripartite motif-containing 11 (TRIM11), and the beta-transducin repeat containing E3 ubiquitin-protein ligase (BTRC)] (Figure 2b, 2c and Table 1),69 which are described in Table 2. All of those genes were upregulated in patients with IDC regarding the controls.
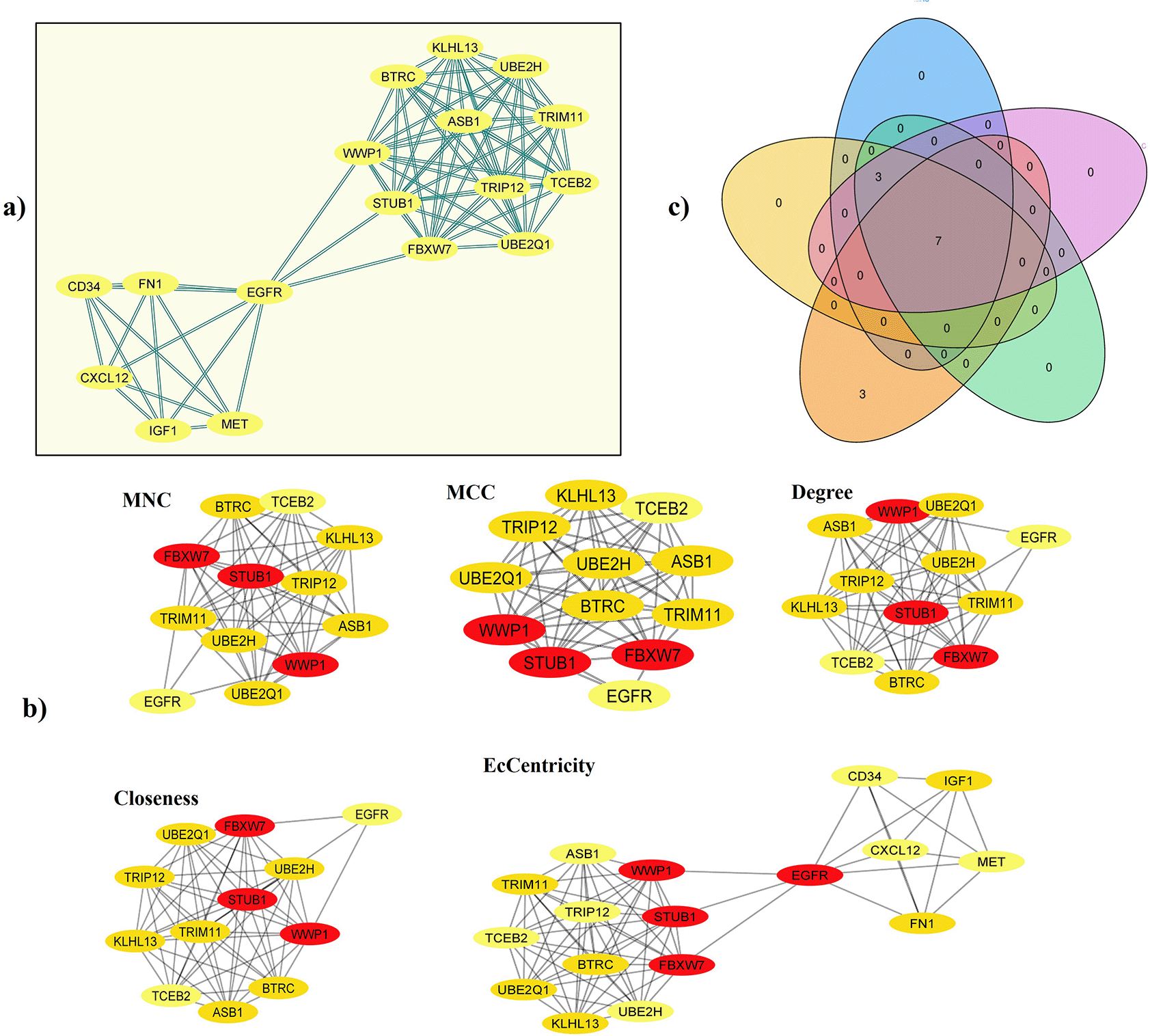
a) Central cluster of the PPI-net built from the 724 common DEGs (MCODE score: 9.125). b) Identification of hub genes (IDC prediction) by cytoHubba algorithms (Degree, EcCentricity, MCC, MNC, and Closeness) in the central cluster of the network. c) Intersection analysis of the genes identified in each algorithm. DEG, differentially expressed gene; PPI, protein-protein interaction; IDC, invasive ductal carcinoma; MCC, Maximal Clique Centrality; MNC, Maximum Neighborhood Component.
| Genes ID | MNC | MCC | Degree | EcCentricity | Closeness |
|---|---|---|---|---|---|
| WWP1 | 11.0 | 3628806 | 11.0 | 0.5 | 13.5 |
| STUB1 | |||||
| FBXW7 | |||||
| KLHL13 | 10.0 | 0.33 | 12.17 | ||
| UBE2Q1 | |||||
| TRIM11 | |||||
| BTRC | |||||
| UBE2H | -- | ||||
| TRIP12 | |||||
| ASB1 | |||||
| EGFR | -- | -- | -- | 0.5 | -- |
| FN1 | 0.33 | ||||
| IGF1 | 0.33 |
| Gene symbol | Gene name | UniProtKB - Id - Function |
|---|---|---|
| WWP1 | WW domain containing E3 ubiquitin protein ligase 1 | Q9H0M0 (WWP1_HUMAN): E3 ubiquitin-protein ligase which accepts ubiquitin from an E2 ubiquitin-conjugating enzyme in the form of a thioester and then directly transfers the ubiquitin to targeted substrates. |
| STUB1 | STIP1 homology and U-box containing protein 1 | Q9UNE7 (CHIP_HUMAN): E3 ubiquitin-protein ligase which targets misfolded chaperone substrates towards proteasomal degradation. Collaborates with ATXN3 in the degradation of misfolded chaperone substrates: ATXN3 restricting the length of ubiquitin chain attached to STUB1/CHIP substrates and preventing further chain extension. |
| FBXW7 | F-box and WD repeat domain containing 7 | Q969H0 (FBXW7_HUMAN): Substrate recognition component of a SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complex which mediates the ubiquitination and subsequent proteasomal degradation of target proteins. |
| KLHL13 | Kelch like family member 13 | Q9P2N7 (KLH13_HUMAN): Substrate-specific adapter of a BCR (BTB-CUL3-RBX1) E3 ubiquitin-protein ligase complex required for mitotic progression and cytokinesis. |
| UBE2Q1 | Ubiquitin conjugating enzyme E2 Q1 | Q7Z7E8 (UB2Q1_HUMAN): Catalyzes the covalent attachment of ubiquitin to other proteins. |
| TRIM11 | Tripartite motif containing 11 | Q96F44 (TRI11_HUMAN): E3 ubiquitin-protein ligase that promotes the degradation of insoluble ubiquitinated proteins, including insoluble PAX6, poly-Gln repeat expanded HTT and poly-Ala repeat expanded ARX. Mediates PAX6 ubiquitination leading to proteasomal degradation, thereby modulating cortical neurogenesis. |
| BTRC | Beta-transducin repeat containing E3 ubiquitin protein ligase | Q9Y297 (FBW1A_HUMAN): Substrate recognition component of a SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complex which mediates the ubiquitination and subsequent proteasomal degradation of target proteins. Recognizes and binds to phosphorylated target proteins. |
WW domain-containing E3 ubiquitin-protein ligase 1 (WWP1), STIP1 homology and U-box containing protein 1 (STUB1), F-box and WD repeat domain containing 7 (FBXW7), kelch like family member 13 (KLHL13), ubiquitin-conjugating enzyme E2 Q1 (UBE2Q1), tripartite motif-containing 11 (TRIM11), beta-transducin repeat containing E3 ubiquitin-protein ligase (BTRC), ubiquitin conjugating enzyme E2 H (UBE2H), thyroid hormone receptor interactor 12 (TRIP12), ankyrin repeat and SOCS box containing 1 (ASB1), epidermal growth factor receptor (EGFR), fibronectin 1 (FN1), insulin-like growth factor 1 (IGF1), Maximum Neighborhood Component (MNC), Maximal Clique Centrality (MCC), protein-protein interaction (PPI).
Enrichment analysis of hub genes was developed in FunRich, classifying them by their ‘biological process’, ‘molecular function’, ‘cellular components’, and the ‘Catalogue of Somatic Mutations in Cancer (COSMIC)’ (RRID:SCR_002260). The results obtained showed that hub genes were mainly enriched in “protein metabolism”, “metabolism”, “cell growth” and “energy pathways”; in turn, among the main molecular functions, analyzed by KEGG pathways showed that hub genes were mainly enriched in “Ubiquitin-specific protease activity”, “cytoskeletal protein binding”, and “ligase activity”; these were associated with the main cellular components where genes are found (“nucleoplasm”, “ubiquitin ligase complex”, “SCF ubiquitin ligase complex”, “ubiquitin conjugating enzyme complex” and “nuclear inclusion body”). Finally, according to COSMIC, the “breast”, “endometrium”, “stomach”, “bone”, and “soft tissue” were the primary site of action of the hub genes (Figure 3).27,28
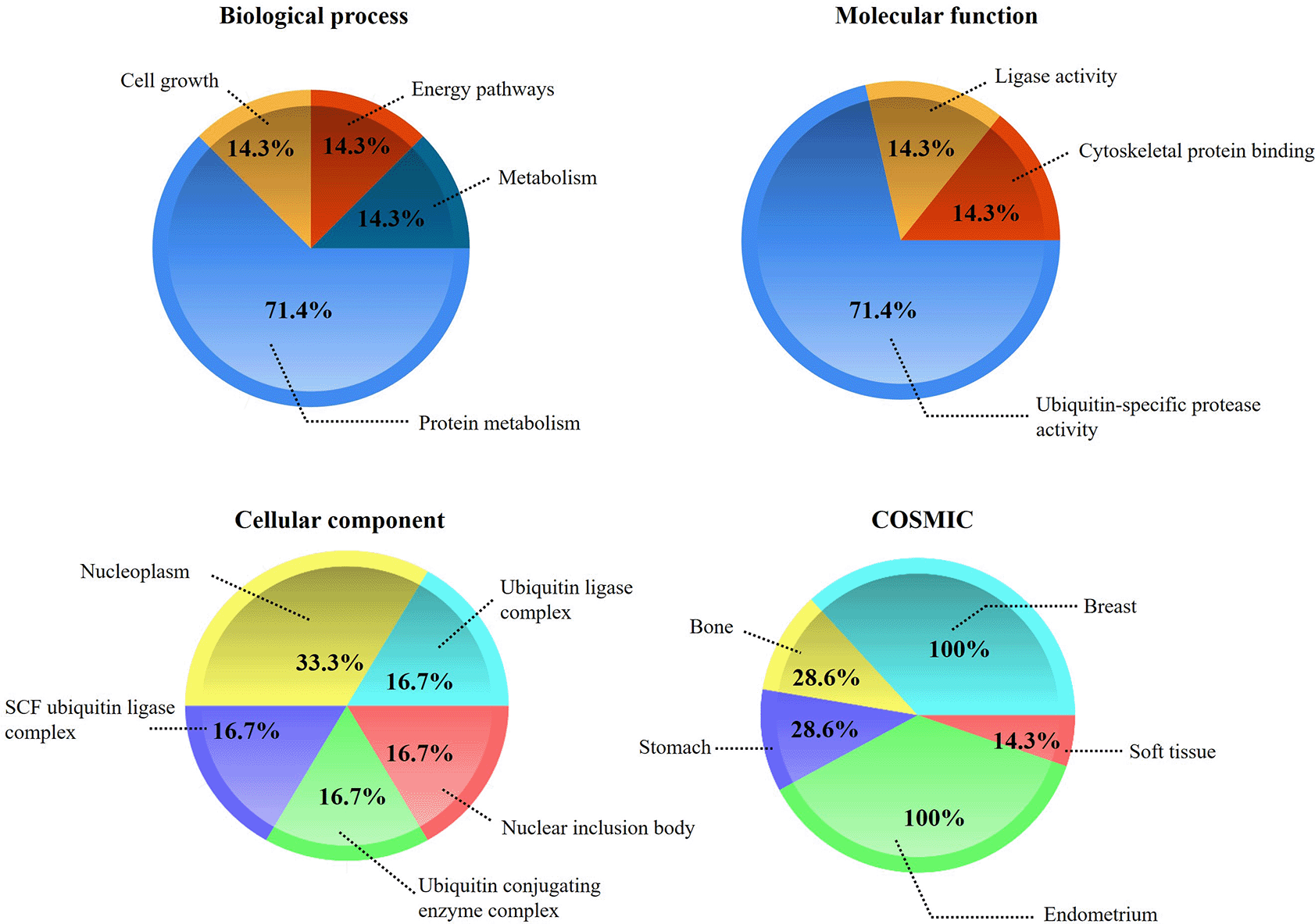
The figure shows the enrichment percentages in terms of ‘biological process’, ‘molecular function’, ‘cellular component’, and the ‘Catalog of Somatic Mutations in Cancer (COSMIC)’. The enrichment analysis was performed in FunRich.
The analysis developed in GEPIA is shown in Figures 4 and 5; this evidenced no statistical differences in expression patterns of hub genes in different stages of IDC. The concentrations of the genes remain constant throughout the evolutionary process of the disease, which could denote an important prognostic factor (Figure 4). On the other hand, high expression of BTRC, FBXW7, and WPP1 was related to the low percentage of survival of the patients (Figure 5).
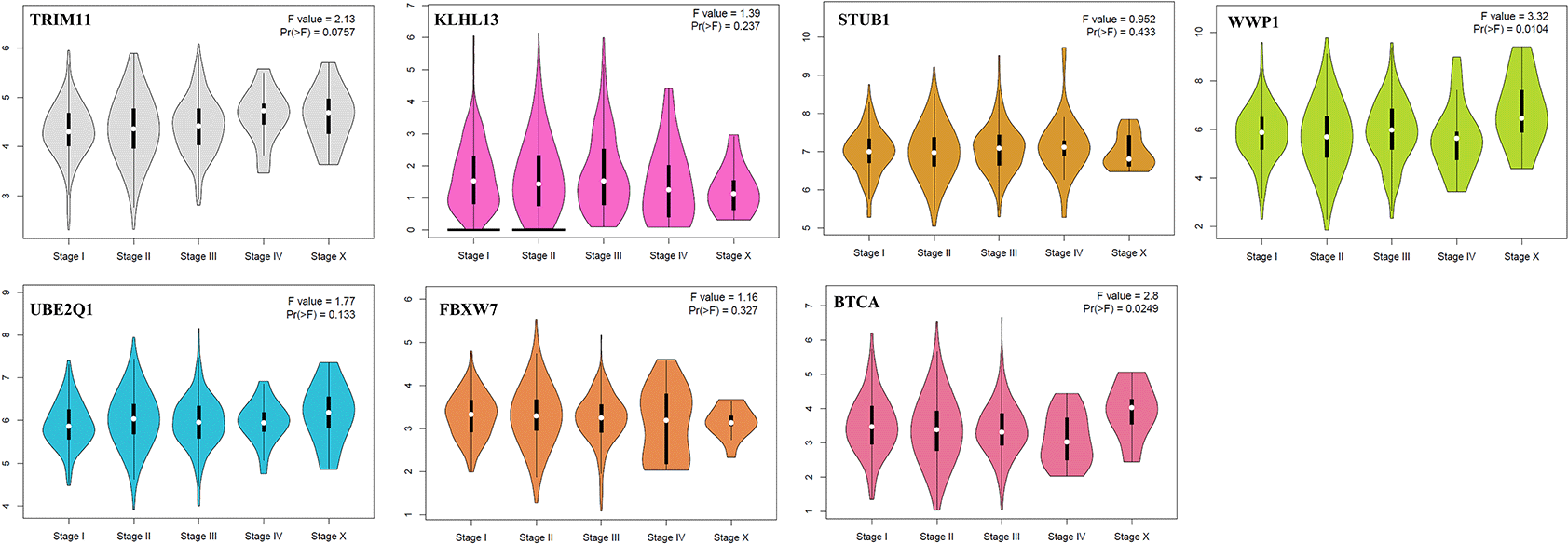
IDC, invasive ductal carcinoma; GEPIA, Gene Expression Profiling Interactive Analysis; WWP1, WW domain-containing E3 ubiquitin-protein ligase 1; STUB1, STIP1 homology and U-box containing protein 1; FBXW7, F-box and WD repeat domain containing 7; KLHL13, kelch like family member 13; UBE2Q1, ubiquitin-conjugating enzyme E2 Q1; TRIM11, tripartite motif-containing 11; BTRC, beta-transducin repeat containing E3 ubiquitin-protein ligase.
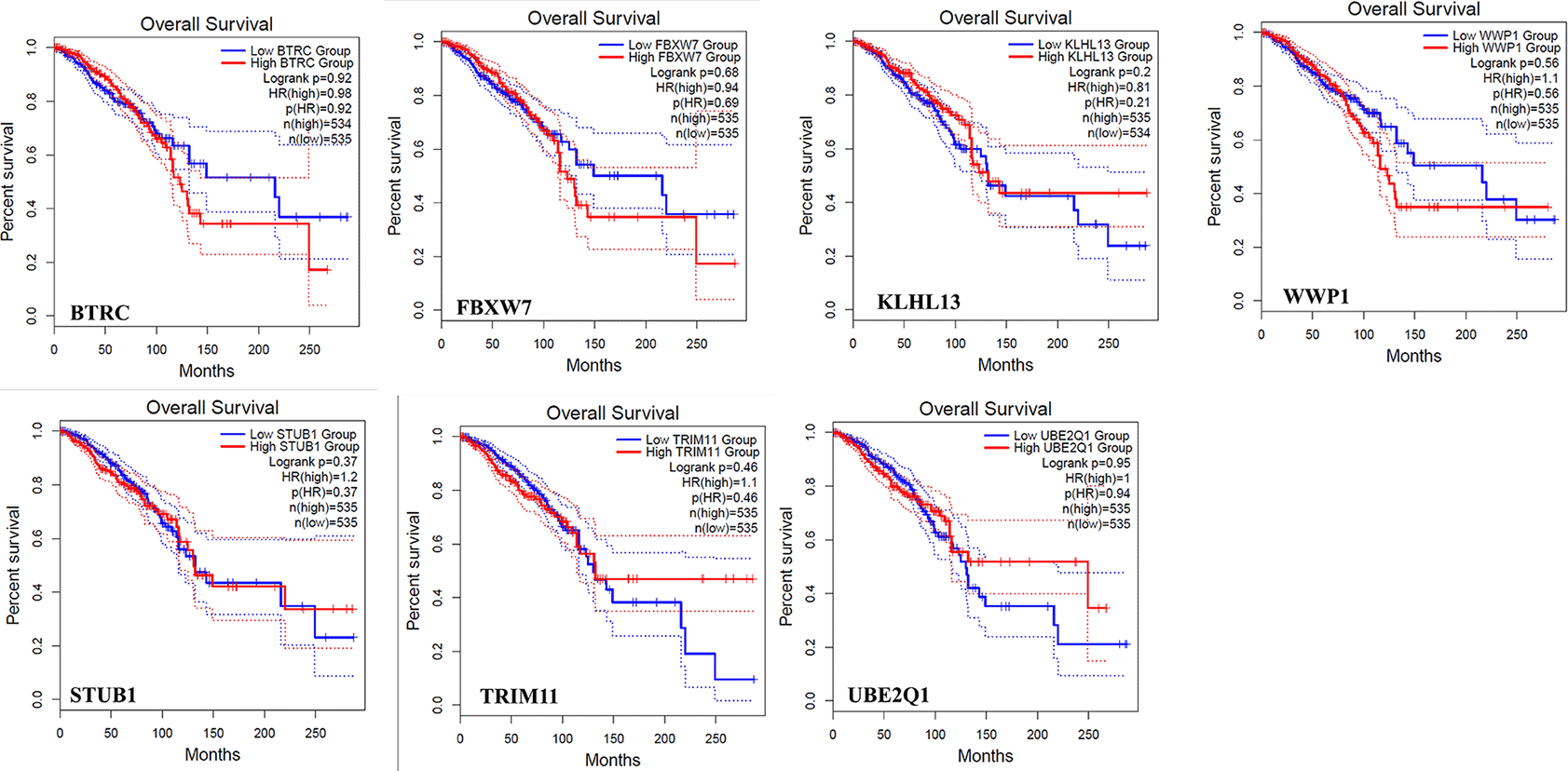
IDC, invasive ductal carcinoma; GEPIA, Gene Expression Profiling Interactive Analysis; WWP1, WW domain-containing E3 ubiquitin-protein ligase 1; STUB1, STIP1 homology and U-box containing protein 1; FBXW7, F-box and WD repeat domain containing 7; KLHL13, kelch like family member 13; UBE2Q1, ubiquitin-conjugating enzyme E2 Q1; TRIM11, tripartite motif-containing 11; BTRC, beta-transducin repeat containing E3 ubiquitin-protein ligase.
IDC is a nonspecific invasive carcinoma that belongs to epithelial tumors. This cancer is considered extremely malignant and the main cause of death in women.30,31 Thus, the search for molecular markers that allow its detection in the early stages is necessary for the diagnosis, early treatment, and prognosis of patients. In this sense, several bioinformatics techniques were integrated into this study, whose objective was to investigate data to screen and identify hub genes related to IDC. Two datasets, GSE29044 and GSE32291, were screened for IDC. Seven gene hubs in common were discovered (WWP1, STUB1, FBXW7, KLHL13, UBE2Q1, TRIM11, and BTRC) (Table 2).
The WWP1 encodes the WW domain-containing E3 ubiquitin-protein ligase 1 protein, a HECT domain-containing E3 ligase regulating apoptosis,32 which has been associated with colorectal, osteosarcomas, oral, gastric, melanoma, prostate, hepatocellular, and BC.33 In BC, WWP1 is frequently amplified and overexpressed34,35; also, the overexpression of WWWP1 in colorectal cancer and BC has been associated with the worst prognosis and poor survival in patients.36 The expression of WWP1 in breast tumors correlated with the positive ER (ERα) status, inducing breast cell growth. On the other hand, WWP1 depletion in ERα positive BC cell lines suppressed cell proliferation and induced apoptosis.32 Despite the above, the role of WWP1 in IDC is not yet well studied. In this context, Chen et al. (2009) reported that WWP1 is overexpressed in a cell line of IDC and was associated with ER and IGF-1R proteins.37,38 On the other hand, Zhou et al. (2012) demonstrated that WWP1 depletion by small interfering (si) RNA activated the extrinsic apoptotic pathway increasing the TRAIL-induced caspase-8 recruitment.32 Although WWP1 has not been associated with any stage of BC, the results of this study show its importance in IDC.
The STUB1 encodes the carboxyl-terminus of HSP70-interacting protein (CHIP); this is a co-chaperone protein that interacts with Hsp70 and negatively regulates chaperone functions. In oral, lung, and colorectal cancer, STUB1 seems to have an important role in the progression of cancer.39,40 According to Lui et al. (2020), the lower expression of STUB1 in oral cancer can be linked to a poorer prognosis.39 Xu et al. (2011) reported that overexpression of STUB1 (CHIP expression) glioma cell lines is associated with the histological grade of the tumor.41 In BC, Kajiro et al. (2009) reported that knockdown of CHIP in breast cancer cells resulted in rapid tumor growth and metastatic phenotypes,42 while Hiyoshi et al. (2014) found that promoting the expression of CHIP can inhibit cell growth and metastatic potential of BC cells.43 Also, Wei et al. (2021) indicated that overexpression of Tripartite motif-containing protein 6 (TRIM6) promoted the degradation of STUB1, which facilitated the growth and migration of malignant cells.44 According to the aforementioned, STUB1 could participate in different ways in the progression of cancer, however, in BC, it may be acting as a tumor suppressor. Regarding the IDC, there is no available evidence associating the presence of the STUB1 with this type of BC.
The TRIM11 encodes the Tripartite motif-containing 11 protein, identified as an oncogene in colon, hepatocellular, and lung cancer. However, its role in BC cells remains unclear. In this sense, Song et al. (2019) reported that TRIM was overexpressed in BC tissues, which was linked to the metabolism of glycolysis.45 Also, Tang et al. (2020) found that the protein level of TRIM11 is highly correlated with ERα, and its depletion significantly decreases the cell proliferation and migration of BC cells.46 As was described above, TRIM6 (member TRIM family), was associated with the degradation of STUB1 on BC cells; thus, we hypothesized that in IDC, these genes could be related metabolically. However, there is no available evidence linking TRIM 11 to invasive ductal cancer.
The FBXW7 encodes the F-box protein family members and has a role important in cell cycle regulation, transcriptional regulation, apoptosis, and cell signal transduction.47,48 The FBXW7 in triple-negative BC (TNBC) has been related to the suppression of proliferation and invasion of TNBC cells. Wu et al. (2020) reported that the inhibition of the TLR4/NF-κB pathway could increase the BXW7 expression, which suppresses the proliferation and invasion of TNBC cells,49 while Singh et al. (2020) demonstrated that downregulation of FBXW7 in a mouse model increased the tumorigenesis and metastasis in TNBC cells.50 Also, Wang et al. (2022) reported that microRNA (miR)-223-3p decreases the expression of FBXW7, which promotes the invasion and metastasis of BC cells.51 According to the studies described above, FBXW7 could have an important role as a suppressor of tumors in BC. Though, its function in IDC has not yet been studied.
The KLHL13 encodes Kelch-like proteins (KLHLs), which act as substrate adaptors of Cullin3-RING ligases (CRL3). CRL3 regulates the degradation of proteins that function as tumor suppressors, which participate in tumor development.52 In this context, Xiang et al. (2021) reported that the upregulation of KLHL proteins contributes to the progression of lung cancer through binding with CRL3, which showed that KLHL13 could be considered a potential target therapeutic.53 However, the involvement of this gene in BC and IDC has not been studied.
The UBE2Q1 encodes ubiquitin-conjugating enzyme E2 Q1 (UBE2Q1), identified as upregulated in human breast and colorectal cancer.54,55 Shafiee et al. (2015) showed that UBE2Q1 in BC cell lines was overexpressed. Also, these authors found that UBE2Q1 could be interacting with p53 through a complex, which explains its involvement in the proliferation and migration of tumor cells.56 Also, Topno et al. (2021) identified UBE2Q1 as a potential prognostic marker in high-grade serous ovarian cancer, using an integrated gene expression analysis and gene co-expression network analysis (WGCNA).57 Nevertheless, the involvement of UBE2Q1 in IDC remains unstudied at present.
The BTRC encodes F-box protein, which has been associated with colorectal, glioma, esophageal, and BC. In this sense, Zheng et al. (2020) reported that the inhibition of BTRC by miR-224 in colorectal cancer promotes cell migration and invasion. The miR-224 silencing promoted the overexpression of BTRC, which decreased the cell progression,58 while Zhou et al. (2021) found that the invasion and migration cells induced by miR-193a-3p in patients with glioma could be reversed by overexpression of BTRC.59 Zhang et al. (2018) indicated that BTRC activity mediated by upregulated tetraspanin 15 (TSPAN15) in esophageal cancer promotes the degradation of phosphorylated (p-)IκBα and triggers NF-κB nuclear translocation and subsequent activation of transcription of several metastasis-related genes [intercellular adhesion molecule 1 (ICAM1, vascular cell adhesion molecule 1 (VCAM1), urokinase-type plasminogen activator (uPA), matrix metallopeptidase 9 (MMP9), tumor necrosis factor α (TNFα), and C-C motif chemokine ligand 2 (CCL2)].60 Lim et al. (2022) found that BTRC acts as an oncogene in TNBC through NF-κB activation (IκBα ubiquitination).61 As described above, the function of BTCR oncogene/gene suppressor could be dependent on cancer type. Therefore, BTRC, like the other identified genes in this study, could be considered a potential therapeutic target and biomarker in IDC.
Code with which R was fed for the preliminary analysis of the data (Intra-group reproducibility):
Figshare: R-Script_GESE32291_STAGE 1. https://doi.org/10.6084/m9.figshare.2041911962
Figshare: R-Script_GESE32291_STAGE 2. https://doi.org/10.6084/m9.figshare.2041916463
Figshare: R-Script_GESE32291_STAGE 3. https://doi.org/10.6084/m9.figshare.2041916764
Figshare: R-Script_GESE29044_STAGE 1. https://doi.org/10.6084/m9.figshare.2041917065
Data are available under the terms of the Creative Commons Zero “No rights reserved” data waiver (CC0 1.0 Public domain dedication).
Figshare: R-Script_GESE29044_STAGE 2. https://doi.org/10.6084/m9.figshare.2041917666
Figshare: R-Script_GESE29044_STAGE 3. https://doi.org/10.6084/m9.figshare.2041917967
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
Raw data derived from differential expression analysis performed in GEO2R for each IDC stage in the datasets:
Figshare: Raw data derived from differential expression analysis performed in GEO2R for each IDC stage in the datasets. https://doi.org/10.6084/m9.figshare.2041920668
Data are available under the terms of the Creative Commons Zero “No rights reserved” data waiver (CC0 1.0 Public domain dedication).
Data derived from the analysis in Cytohubba from Cytoscape:
Figshare: Data derived from the analysis in Cytohubba from Cytoscape. https://doi.org/10.6084/m9.figshare.2041921869
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
Figshare: Supplementary Figure S1. https://doi.org/10.6084/m9.figshare.2029384270
Figshare: Supplementary Figure S2. https://doi.org/10.6084/m9.figshare.2029384571
Figshare: Supplementary Figure S3. https://doi.org/10.6084/m9.figshare.2029384872
Data are available under the terms of the Creative Commons Zero “No rights reserved” data waiver (CC0 1.0 Public domain dedication).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)