Keywords
Invasive ductal carcinoma, prognostic biomarkers, hub genes, microarray technology, differentially expressed genes, GEO, GEO2R
This article is included in the Oncology gateway.
This article is included in the Bioinformatics gateway.
This article is included in the Bioinformatics in Cancer Research collection.
Invasive ductal carcinoma, prognostic biomarkers, hub genes, microarray technology, differentially expressed genes, GEO, GEO2R
The revised edition of the research article titled "Identification of prognostic biomarkers of invasive ductal carcinoma by an integrated bioinformatics approach" encompasses significant improvements to the findings. These modifications are a result of expanding the initial analysis and incorporating a new dataset, in accordance with the annotations and recommendations provided by the reviewers. The outcome entails enhanced performance, characterized by increased traction and bolstered backing for the suggested hub genes.
See the authors' detailed response to the review by Xingxin Pan
See the authors' detailed response to the review by Russell Hamilton
Breast cancer (BC) is the most prevalent and frequently diagnosed neoplasm in women around the world,1–3 except in West Africa, where cervical cancer is more prevalent.4 According to The Global Cancer Observatory, in 2020, there were more than 2.3 million new cases of BC worldwide, being the fifth cause of death related to cancer. In general, BC incidence and mortality rates have increased over the last three decades; in fact, by 2030, the global number of newly diagnosed cases is expected to reach 2.7 million annually, along with 0.87 million deaths; this figure could be higher in low- and middle-income countries due to several factors that include the development of health systems.5
Several authors converge that BC is a heterogeneous pathological condition (at the histological and molecular level) that can be categorized as a collection of several diseases with different risk factors, clinical manifestations, pathological characteristics, and therapeutic responses.6–8 Currently, high-throughput techniques like microarrays may be used to identify BC genetic profiles, which is essential for categorizing them. In 2000, Perou et al. conducted a statistical analysis of estrogen receptor (ER) and human epidermal growth factor receptor 2 (HER2) gene expression data to develop a molecular classification of BC into four distinct groups: i) luminal-A [ER(+) - HER2(-)], ii) luminal-B [ER(+) - HER2(+)], iii) HER2-enriched [ER(-) - HER2(+)), and iv) basal-type [ER(-) - HER2(-)].9
Clinical therapy for BC changed as a result of Perou et al.’s categorization, moving away from methods oriented on tumor burden and toward approaches centered on biology. A surrogate categorization of five groups based on histology and genetic traits is now used in clinical practice. These groups include hormone receptor-positive BCs that express the ER and/or progesterone receptor (PR), whereas triple-negative BCs do not express the ER, PR, or HER2 (TNBC).6
The histological analysis of BC considers their anatomical origin, classifying them primarily as carcinomas and sarcomas. The first type comes from epithelial cells of the breast (lobes and terminal lactiferous ducts responsible for milk) or from underlying mammary stem cells, while sarcomas originate in connective tissues, such as blood vessels and myofibroblasts, which support the ducts and lobules. BC are further divided by their great heterogeneity into in situ (found in their main lobules and ducts) and invasive carcinomas, which can penetrate nearby tissues and, if left unchecked, may spread to other tissues and organs of the breast body.8 Invasive carcinomas are further divided into morphologically identifiable types and “not otherwise specified” (NOS) or “no special type” (NST) types, based on their morphology;4,8 of all of them, NST invasive ductal carcinoma (IDC) represents the most common type of invasive carcinoma (50% to 80% of newly diagnosed BC cases), followed by invasive lobular carcinoma.3,5,10,11
IDC is a diverse collection of tumors that develop in the cells that line the milk ducts in the breast and then spread to the tissues and walls of the duct. These may also have the ability to spread (metastasize) to nearby areas of the body through the lymphatic circulation.12,13 Under the microscope, IDCs display a wide range of histopathologic traits; they take the shape of diffuse sheets, well-defined nests, cords, or single cells, and their tubular differentiation is frequently well-developed, hardly noticeable, or completely absent.10
The clinicopathological characteristics of IDCs have been well described in the literature,12 and the discovery of diagnostic and prognostic biomarkers for the many subtypes of BCs has been essential to their understanding and management.14 These prognostic factors are primarily based on tumor stage, hormone receptor status (ER and PR), nuclear grade, and Her-2/neu expression.3 Around 70–75% of invasive breast carcinomas express ER and PR, which indicate a favorable prognosis, a less aggressive form of the disease, and a higher rate of patient survival overall,15,16 while the detection probability of metastatic or recurrent BC rises from 50% to even more than 80% with HER2 overexpression, thus constituting one of the first stages of breast carcinogenesis.5
In addition to the aforementioned, the high penetrance genes BRCA1 (breast cancer 1, located on chromosome 17) and BRCA2 (breast cancer 2, located on chromosome 13) are also primarily related to the increased risk of breast carcinogenesis (responsible for 90% of hereditary BC cases); they have been described as important in the development of BC. BRCA1 and BRCA2 are tumor suppressor genes with a well-defined role in DNA repair; mutations in either gene increase the risk.17
Sporadic mutations of genes such as TP53 (transcription factor involved in cell cycle arrest, cellular senescence, apoptosis, metabolism, DNA repair, and other functions),18 PIK3CA (Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha), which participates in cell growth and division, cell survival, and cell movement, among others,19 MYC that encodes for the c-MYC protein, which participates in cellular differentiation, apoptosis, growth, replication, and metabolism, overexpression of c-MYC in BCs indicates a malignant and invasive phenotype with early recurrence,20,21 PTEN (Phosphatase And Tensin Homolog), an inhibitor of protein kinase B (Akt) and PI3K signaling, essential for cell growth, survival, and cell cycle progression,22,23 CCND1 (key regulator of the cell cycle that mediates the transition from G1 to S phase, its overexpression has been found in 50-70% of BC),24,25 and STK1 (Thymidine kinase a fundamental enzyme in DNA synthesis and cellular proliferation, this is associated with poorer prognosis in many cancer types, including breast),26 are also reported as key for BC prognosis, whose carriers are more susceptible to BC since they are genes that mostly modulate the cell cycle and whose mutations promote maintaining proliferation and/or inhibiting apoptosis.
On the other hand, mutations in DNA repair genes have also been reported that can interact with the BRCA genes (ATM, PALB2, BRIP1, or CHEK2), which are involved in the induction of mammary carcinogenesis; however, they are characterized by lower penetrance compared to BRCA1 or BRCA2.5 Finally, the Ki-67 protein and E-cadherin are currently also prognostic biomarkers for BC20; Ki-67 is an excellent marker of cell proliferation, whose proliferation index Mib1 (antibody against Ki-67) remains a reliable diagnostic biomarker of BC (proliferative activities determined by Ki-67 reflect the aggressiveness of the cancer together with the response to treatment and time to recurrence). E-cadherin, for its part, is used as a predictor of tumor size, stage, or the state of the lymph nodes.5,6
Early diagnosis and proper treatment of IDCs are extremely important for patient survival and a good prognosis. IDCs tend to significantly affect the physical and psychological health of young women, given their tendency to cause lymphatic metastasis and recurrence in advanced patients and their influence on the appearance of the breasts after surgical resection. For this reason, clinical practice seeks every day to promote individualized treatment; therefore, the search for potential prognostic biomarkers, target molecules, and signaling pathways that may be associated with the disease is considered an important step toward achieving.27,28 Prognostic markers are also helpful in determining the effectiveness of the established intervention (surgical or pharmacological treatment), the probability of recurrence, and the establishment of additional follow-up and treatment strategies.29
Due to BC’s heterogeneity and high capacity for metastasis, despite efforts to identify biomarkers and the variety of genetic factors currently accepted, an increasing number of patients are demanding personalized treatments, which necessitates the development of novel biomarkers for diagnosis and prognosis that allow for an early evaluation of the development of the pathology to formulate effective diagnosis and treatment strategies.30–32
Nowadays, the analysis of gene expression profiles [verification of differentially expressed genes (DEGs)] using bioinformatics tools has represented a notable advance in research in clinical oncology aimed at the identification of genes related to tumors, new molecular markers of diagnosis and prognosis, and evaluation of therapeutic effects, among others.33 DEGs and protein-protein interaction network (PPI-net) analysis have been widely used to identify biomarkers and potential drug targets. Open-access databases such as Gene Expression Omnibus (GEO) are broadly employed as microarray resources for this purpose.34
Previous studies have identified prognostic genes from ductal carcinoma in situ (DCIS), such as fibroblast growth factor 2 (FGF2), growth arrest-specific protein 1 (GAS1), and secreted frizzled-related protein 1 (SFRP1) using GEO35; however, currently, IDC is little understood from the genomic point of view, and there are no studies on the analysis of genes expression using bioinformatic methods. Thus, this study aimed to identify new candidates genes for the prognostic of this type of BC using an integrated bioinformatics approach.
Using the Gene Expression Omnibus, GEO (RRID:SCR_005012) web server (https://www.ncbi.nlm.nih.gov/geo/), the transcriptomic data collections were selected. GEO is an open database comprising microarray and sequencing data contributed by the scientific community.36 The datasets were selected using the MeSH (RRID:SCR_004750) terms (“carcinoma, ductal, breast” [MeSH Terms] OR invasive ductal carcinoma [All Fields]) AND “Homo sapiens”[porgn]). Following are the inclusion criteria used to identify the datasets: i) data derived from human studies; ii) collections that evaluated the initial phases of the IDC; iii) stable and complication-free health condition of enrolled patients; and iv) array-analyzed collections.
To verify the intra-group data repeatability per each group of datasets, as proposed by Xu et al. (2019), for it, we developed a Pearson’s correlation test and a principal component analysis (PCA) using the R programming language, R Project for Statistical Computing (RRID:SCR_001905). The degree of correlations between all samples from the same dataset was visualized by heat maps built in R.37
DEGs were screened out from the first three stages of IDC through online analysis in GEO2R (RRID:SCR_016569), which is an interactive online tool from GEO that finds DEGs by comparing the original submitter-supplied processed data tables using the GEOquery (RRID:SCR_000146) and limma R packages from the Bioconductor project.38,39 Initially, the experimental groups were built from the datasets, grouping the samples as tissues with IDC and controls (adjacent disease-free tissues). A comparative analysis was carried out for each IDC stage evaluated. The cut-off criterion was P < 0.05 and a fold-change among ≥ 1.5 or ≤ -1.5. Volcano plots with the DEGs found were drawn in GEO2R. An intersection analysis between DEGs extracted from the three datasets was made using Venn diagrams delineated in the functional enrichment analysis tool (FunRich).37,40 DEGs with log FC < 0 was regarded as down-regulated genes, and vice versa.41
A protein-protein interaction network (PPI-net) was built using the online Search Tool for the Retrieval of Interacting Genes [STRING (RRID:SCR_005223)]; for this, we used the genes generated from the intersection analysis between the selected DEGs (up- and down-regulated genes). The PPI-net was visualized in Cytoscape (RRID:SCR_003032) (version 3.8.0),42 and through the MCODE App (RRID:SCR_015828) (Molecular complex detection tool; version 1.6.1) was identified the most important module of the network using as criteria a degree of cut-off of 2, scores >5, a maximum depth equal to 100, a node score cut-off of 0.2, and a k-score of 2. Genes with degrees ≥10 were selected as hub genes.37,38
After the identification of the main module of the network, the top 10 central genes were evaluated through the cytoHubba (RRID:SCR_017677) application of Cytoscape,43 using the ten most reported calculation methods: Degree, EcCentricity, Edge Percolated Component (EPC), Closeness, Maximum Neighborhood Component (MNC), Maximal Clique Centrality (MCC), Betweenness, Radiality, BottleNeck, and Stress.44 Overlapping of 10 algorithms were carried out using the “UpSetR” package (RRID:SCR_022731).45
Functional enrichment analysis of the hub genes identified was performed in the DAVID tool (RRID:SCR_001881) through the Kyoto Encyclopedia of Genes and Genomes (KEGG) (RRID:SCR_012773). We analyzed the enrichment around the molecular function, biological process, cellular component, and pathways analysis.46 On the other hand, the expression patterns of hub genes between different stages of IDC were analyzed based on Gene Expression Profiling Interactive Analysis (GEPIA) (RRID:SCR_018294), a web server for cancer and normal gene expression profiling and interactive analyses.47
We examined the relationship between overall survival in BC and the expression of core genes using the Kaplan-Meier plotter database (RRID:SCR_018753). On the other hand, Breast Cancer Gene-Expression Miner v4.9 (bc-GenExMiner) was used to assess the interaction between the hub genes. Finally, mRNA-seq data for BC was retrieved from the Cancer Genome Atlas (TCGA) database (RRID:SCR_003193) to confirm the significance of the hub genes.33
All analyses were conducted in GraphPad Prism (RRID:SCR_002798) (version 8.0.2) [free alternative, JASP (RRID:SCR_015823) (version 16.3)] and RStudio (RRID:SCR_000432). One-way analysis of variance (ANOVA) was used for comparing the mean between groups in the analyses conducted in GEPIA. P<0.05 was considered to indicate a statistically significant difference.
Three gene expression profiling datasets GSE29044,48 GSE32291, and GSE2142249 were downloaded GEO, which were based on the platforms [GPL570 (HG-U133_Plus_2 - Affymetrix Human Genome U133 Plus 2.0 Array)], [GPL4091 (Agilent-014693 Human Genome CGH Microarray 244A (Feature number version)], and [GPL570 (HG-U133_Plus_2 - Affymetrix Human Genome U133 Plus 2.0 Array)], respectively.
GSE29044 was a collection that analyzed the whole-genome mRNA expression profile from 73 patients with tumors and 36 adjacent disease-free tissues using the Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays.50 Using Agilent’s whole-genome CGH arrays. GSE32291 analyzed 394 invasive ductal breast cancers and 20 normal breast biopsies; while the GSE21422 analyzed five non-malignant control (healthy tissue samples), nine DCIS and five invasive ductal carcinomas from patients with breast reduction surgery.49 Clinical details of samples utilized from each dataset are condensed in the supplemental material (Supplementary Tables S1–S3).
The features of selected datasets are shown on supplementary tables, S1–S3 which can be found as Underlying data. Samples in the dataset were chosen based on the following inclusion criteria: that they be a wild-type strain and both ER and PR positive. R script for GSE29044, GSE32291 and GSE21422 can be found on GitHub (Extended data). Pearson’s correlation coefficient showed that datasets had a strong correlation among the samples from the control group and IDC (Supplementary Figures S1–S3, which can be found as Extended data). On the other hand, the PCA classified the samples of the dataset into two components, indicating that the repeatability of the data was adequate. In the PC2 dimension, the distances between samples from the control group and the IDC of GSE32291 dataset were close, while they were close for samples from GSE329044 and GSE21422 in both dimensions.
DEGs screened out from the datasets were analyzed through the construction of volcano plots in GEO2R. Nodes that conformed to the cut-off criterion (fold-change ≥1.5 or ≤-1.5, and a P<0.05) were represented in blue or red color, and classified as downregulated and upregulated DEGs regarding the control, respectively (Supplementary Tables S4–S6 - Underlying data). An intersection analysis was made in FunRich with downregulated and upregulated DEGs from each dataset, resulting in 497 and 495 common DEGs, respectively (Supplementary Table S7 - Underlying data) (P<0.05) (Figure 1).
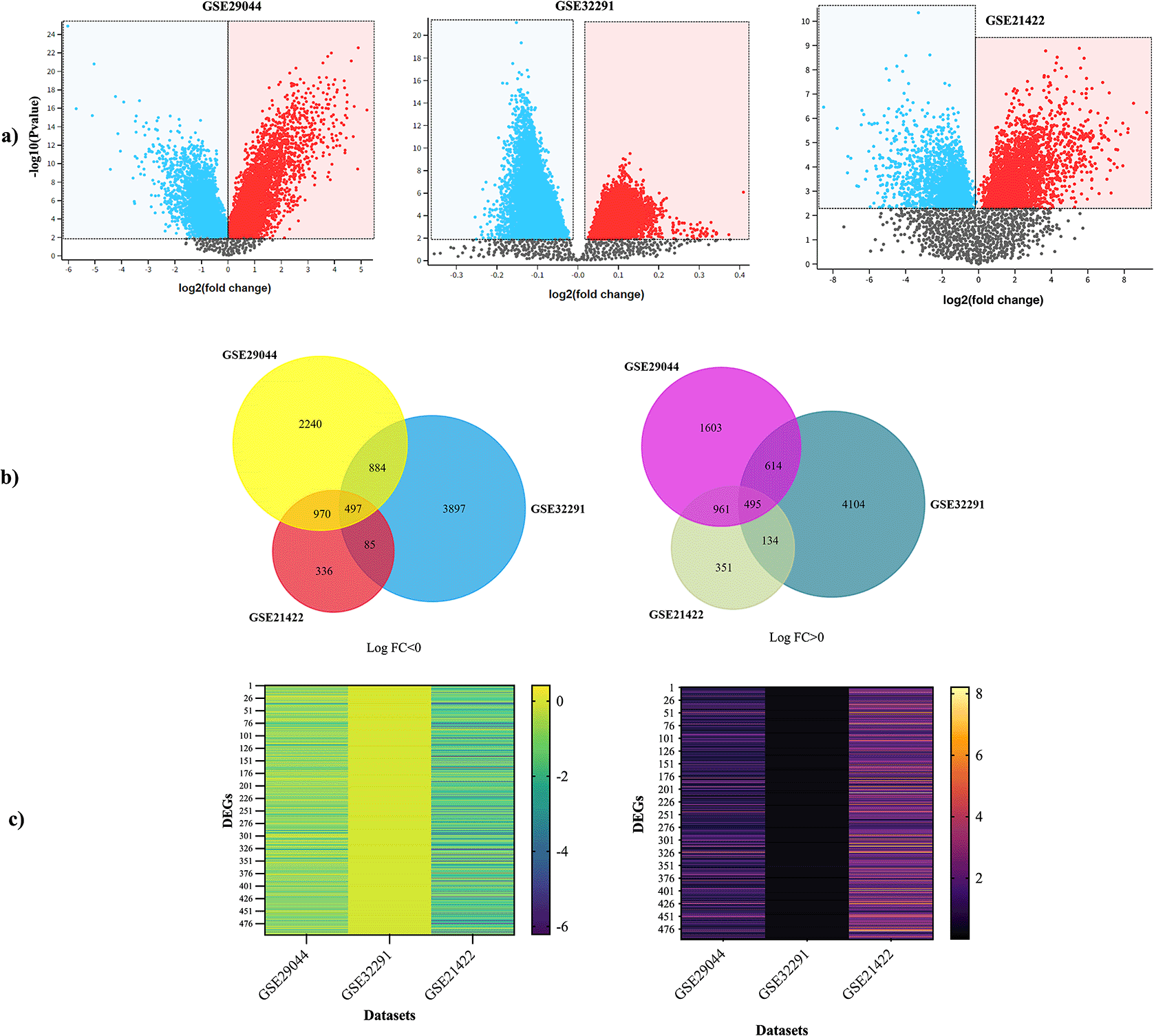
a) Volcano plots obtained in GEO2R show the difference in gene expression between tissues of IDC and controls. The X and Y-axis represent the fold-change and the P-value (log-scaled). Each symbol represents a different gene; red and blue symbols represent upregulated and downregulated genes. b) Venn diagram showing the shared up and down-regulated genes per stages assayed. c) Heatmap showed the common DEGs in three datasets. Left heatmap indicated the common down-regulated DEGs, and right heatmap indicated the common up-regulated DEGs. DEG, differentially expressed genes; IDC, invasive ductal carcinoma.
From the 992 common DEGs, a PPI network was built in STRING using the following parameters: medium confidence of > 0.4 (minimum required interaction score) and that the network will only display the query proteins; Supplementary Figure S3, which can be found as Extended data described the network features. Next, we identified the most significant PPI-net module by the MCODE app from Cytoscape, which had 375 edges, 29 nodes, and a score of 26.786; from it we identified the 19 principal genes of the PPI-net classified by 10 different cytohubba algorithms (Degree, EcCentricity, EPC, Closeness, MNC, MCC, Betweenness, Radiality, BottleNeck, and Stress), depending on the level of categorization of the algorithms, the seven center hub genes were chosen from this group, KIF23, and ASPM (classified by all algorithms); and HMMR, PRC1, AURKA, RACGAP1, CENPF (classified by nine algorithms) (Figure 2 – Table 1). Details of these hub genes are shown in Table 2.

a) Central cluster of the PPI-net built from the 992 common DEGs (MCODE score: 26.786). The other part of the figure shows the classified central genes by the algorithms Betweenness, Stress, Radiality, Closeness, EcCentricity, Degree, MNC, MCC (b), EPC (c), and BottleNeck (d). e) UpSet diagram derived from the analysis of the PPI-net for the identification of hub genes from the superposition of the results of the 10 topological algorithms used (Degree, EcCentricity, EPC, Closeness, MNC, MCC, Betweenness, Radiality, BottleNeck, and Stress). f) PPI-net of hub genes. DEG, differentially expressed gene; PPI, protein-protein interaction; IDC, invasive ductal carcinoma; MCC, Maximal Clique Centrality; MNC, Maximum Neighborhood Component.
Top 7 genes found in the PPI network using the most used ten calculation methods of cytoHubba (MCC, MNC, EPC, Degree, EcCentricity, Closeness, Betweenness, Stress, Radiality, and BottleNeck) from Cytoscape.
| Gene ID | MCC | MNC | Degree | EPC | BN | EC | Clos. | Rad. | Betw. | Stress |
|---|---|---|---|---|---|---|---|---|---|---|
| KIF23 | 1.2 E37 | 28 | 1.2 E16 | 2.0 | 1.0 | 28 | 2 E16 | 3 E15 | 62 | |
| ASPM | ||||||||||
| AURKA | -- | |||||||||
| RACGAP1 | ||||||||||
| CENPF | ||||||||||
| HMMR | -- | 1.0 | ||||||||
| PRC1 | ||||||||||
Function of the ten dentified hub genes.
| Gene symbol | Gene name | UniProtKB - Id - Function |
|---|---|---|
| KIF23 | Kinesin-like protein KIF23 | Q02241 · KIF23_HUMAN: Component of the central spindling complex that functions as a microtubule-dependent and Rho-mediated signaling necessary for the production of myosin contractile rings during the cell cycle. Rho-mediated signaling is essential for cytokinesis. |
| ASPM | Abnormal spindle-like microcephaly-associated protein | Q8IZT6 · ASPM_HUMAN: Regulates the mitotic spindle and coordinates the mitotic processes. The interaction with the katanin complex composed of KATNA1 and KATNB1 appears to have a role in controlling microtubule dynamics at spindle poles, including spindle orientation, astral microtubule density, and poleward microtubule flow. |
| AURKA | Aurora kinase A | O14965 · AURKA_HUMAN: Mitotic serine/threonine kinase that contributes to the regulation of cell cycle progression. |
| RACGAP1 | Rac GTPase-activating protein 1 | Q9H0H5 · RGAP1_HUMAN: Component of the centralspindlin complex that functions as a microtubule-dependent and Rho-mediated signaling necessary for the production of myosin contractile rings during the cell cycle. |
| CENPF | Centromere protein F | P49454 · CENPF_HUMAN: Necessary for chromosomal segregation and kinetochore operation in mitosis. Dynein, LIS1, NDE1, and NDEL1 are necessary for the localization of the kinetochore. By serving as a bridge between recycling vesicles and the microtubule network through its connection with STX4 and SNAP25, it controls the recycling of the plasma membrane. |
| HMMR | Hyaluronan mediated motility receptor | O75330 · HMMR_HUMAN: Receptor for hyaluronic acid. |
| PRC1 | Protein regulator of cytokinesis 1 | O43663 · PRC1_HUMAN: Important cytokinesis regulator that crosses antiparallel microtubules at a 35 nM average distance. Controlling this is necessary for effective cytokinesis and the spatiotemporal development of the midzone. |
Enrichment analysis of hub genes was developed in DAVID tool through KEGG, classifying them by their ‘biological process’, ‘molecular function’, and ‘cellular components’. The results obtained showed that hub genes were mainly enriched in “nuclear division”, “organelle fission”, “mitotic nuclear division” and “nuclear chromosome segregation”; in turn, among the main molecular functions, analyzed by KEGG pathways showed that hub genes were mainly enriched in “microtubule binding”, and “tubulin binding”; these were associated with the main cellular components where genes are found (“mitotic spindle”, “spindle”, “microtubule”, and “spindle pole”). Finally, according to COSMIC, the “breast”, “endometrium”, “ovary”, “skin”, and “soft tissue” were the primary site of action of the hub genes (Figure 3).
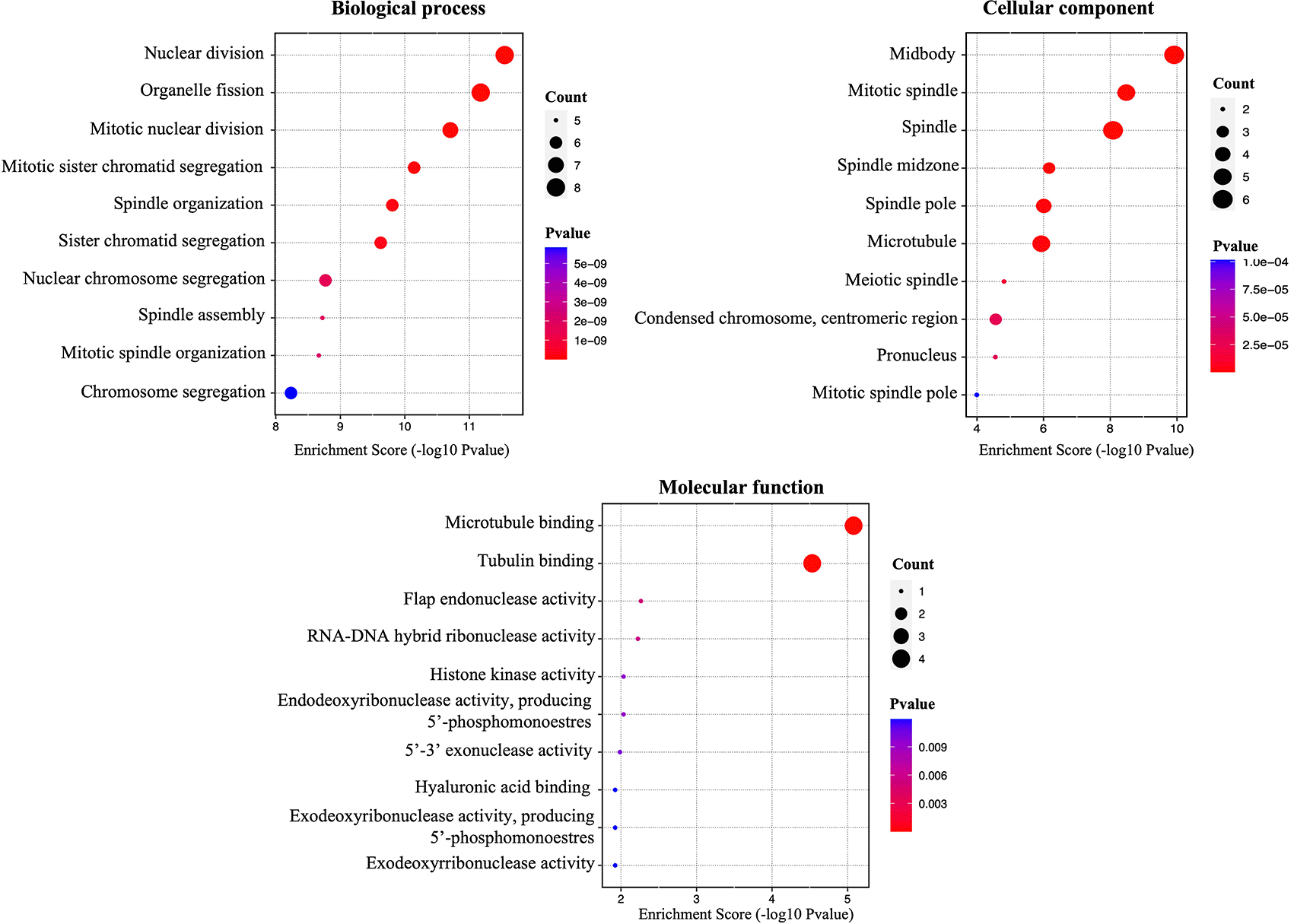
The figure shows the enrichment percentages in terms of ‘biological process’, ‘molecular function’, ‘cellular component’.
Next, we developed a comparative analysis of hub gene expression in GEPIA, which is shown in the Figure 4; this evidenced no statistical differences in expression patterns of hub genes in different stages of IDC. The concentrations of the genes remain constant throughout the evolutionary process of the disease, which could denote an important prognostic factor. On the other hand, all the identified hub genes were associated with poor overall survival in IDC patients (P < 0.05), as indicated by the Kaplan-Meier tracer, high expression of all hub genes was associated with decreased relapse-free survival in patients with breast cancer (P < 1 ×10-16), indicating their significance and positive correlation with overall survival in IDC (Figure 5). In fact, all those genes were up-regulated in patients with IDC regarding the controls (Supplementary table S7 - Underlying data), and besides, there was a strong positive correlation between them (all P < 0.05) (Figure 6). Finally, the significance of the hub genes was then evaluated using mRNA-seq data for BC obtained from the The Cancer Genome Atlas (TCGA). Figure 7 depicts the distribution of mutations in identified hub genes on IDC. ASPM and CENPF were the most frequently mutated genes, accounting for 8.7 and 6.21% of all mutations among 8,667 patients.

IDC, invasive ductal carcinoma; GEPIA, Gene Expression Profiling Interactive Analysis; KIF23, Kinesin-like protein KIF23; ASPM, Abnormal spindle-like microcephaly-associated protein; AURKA, Aurora kinase A; RACGAP1, Rac GTPase-activating protein1; CENPF, Centromere protein F; HMMR, Hyaluronan mediated motility receptor; PRC1, Protein regulator of cytokinesis 1.

Prognostic value of the seven hub genes in IDC patients based on the Kaplan-Meier Plotter. KIF23, Kinesin-like protein KIF23; ASPM, Abnormal spindle-like microcephaly-associated protein; AURKA, Aurora kinase A; RACGAP1, Rac GTPase-activating protein1; CENPF, Centromere protein F; EXO1, Exonuclease 1; HMMR, Hyaluronan mediated motility receptor; PRC1, Protein regulator of cytokinesis 1; NUF2, kinetochore protein nuf2; UBE2C, Ubiquitin-conjugating enzyme E2C.

Cualitative (a) and quantitative analysis (b) of the interrelation expression of seven hub genes performed by bc-GenExMiner software. KIF23, Kinesin-like protein KIF23; ASPM, Abnormal spindle-like microcephaly-associated protein; AURKA, Aurora kinase A; RACGAP1, Rac GTPase-activating protein1; CENPF, Centromere protein F; HMMR, Hyaluronan mediated motility receptor; PRC1, Protein regulator of cytokinesis 1.

KIF23, Kinesin-like protein KIF23; ASPM, Abnormal spindle-like microcephaly-associated protein; AURKA, Aurora kinase A; RACGAP1, Rac GTPase-activating protein1; CENPF, Centromere protein F; EXO1, Exonuclease 1; HMMR, Hyaluronan mediated motility receptor; PRC1, Protein regulator of cytokinesis 1; NUF2, kinetochore protein nuf2; UBE2C, Ubiquitin-conjugating enzyme E2C.
IDC is a nonspecific invasive carcinoma that belongs to epithelial tumors. This cancer is considered extremely malignant and the main cause of death in women.51,52 Thus, the search for molecular biomarkers that allow its detection in the early stages is necessary for the diagnosis, early treatment, and prognosis of patients.
This research employed several bioinformatics tools with the aim of examining data and identifying key genes associated with IDC in three datasets (GSE29044, GSE32291, and GSE21422). As a result, seven hub genes were found, that were up-expressed in all IDC tissue samples (KIF23, ASPM, AURKA, RACGAP1, CENPF, HMMR, and PRC1).
KIF23 and ASPM were identified by all cytohubba algorithms assayed; the first is also known as MKLP1. It is a member of the kinesin superfamily of motor proteins, a group of microtubule-associated motility proteins with multiple functions in normal cellular biological processes, such as cytoplasm separation in mitosis and cytokinesis, transport of vesicles, organelles, chromosomes, and RNA-binding proteins in cells; its globular apex possesses ATPase activity, which can generate energy by hydrolyzing ATP and altering its configuration.53 KIF23 is essential for the formation of the central spindle midbody, and its absence results in cytokinesis defects that lead to the formation of bi- and multinucleated cells.53–55 Several studies have demonstrated that KIF23 plays a critical role in tumorigenesis and cancer progression through the disruption of normal cytokinesis and centrosome formation, resulting in cell division arrest or abnormalities that contribute to the formation of aneuploid cells that promote tumorigenesis. High levels of KIF23 in tumor-affected tissues ere associated with a poor prognosis and are correlated with TNM stage56–58 and recurrence in numerous tumors, including gastric, esophageal, liver, colorectal, pancreatic,57,59,60 cervical, prostate, ovarian, bladder,60,61 lung, gliomas,62 lymphoma, melanoma, and in BC.60,63
KIF23 promotes cancer cell proliferation via direct competitive interaction with the membrane recruitment protein 1 (Amer1), blocking the association of this protein with the adenomatous polyposis coli (APC), thereby relocating Amer1 from the membrane and cytoplasm to the nucleus and attenuating its ability to negatively regulate Wnt/β-catenin signaling, thus activating this signaling pathway. This mechanism has been widely described in gastric cancer (CG); in fact, in vitro and in vivo inhibition of KIF23 by KIF23 knockout mice inhibited not only GC cell proliferation (downregulation of Ki67 and PCNA expression) and tumorigenesis but also migration and invasion and arrested the cell cycle in the G2/M phase.57,59
The Wnt/β-catenin signaling pathway plays a crucial role in cell development and differentiation, and is intimately associated with tumorigenesis, invasion, and metastasis. Additionally, aberrant Wnt/β-catenin signaling is closely associated with a high incidence of numerous human malignancies.64
As far as we are concerned, although there is some existing scientific research that associates the up-regulation of KIF23 in BC tissues, there aren’t any similar works focused on assessing the expression of this gene in IDC samples, which reinforces the analysis made in the present work. KIF23 could similarly activate the Wnt/β-catenin pathway in BC as it does in GC. According to Li et al. (2022), this activation can facilitate the progression of the epithelial-to-mesenchymal transition (EMT). Thus, epithelial cells acquire the morphology of mesenchymal stromal cells and acquire motility and invasiveness. EMT enhances cancer cell migration and invasiveness, thus improving their metastatic potential.65
EMT process has been implicated in the tumorigenic progression of various malignancies, including TNBC, as evidenced by the study conducted by Li et al. (2022). The authors employed microarray analysis of publicly available datasets (GSE41313, GSE5460, and GSE1456) as well as The Cancer Genome Atlas (TCGA) data to investigate the expression of KIF23 in BC tissues and cell lines. Additionally, they conducted in vitro and in vivo functional experiments to assess the impact of KIF23 on tumor growth and metastasis in TNBC. The results revealed a significant upregulation of KIF23 expression in TNBC cell lines compared to luminal cell lines. Consequently, their research findings indicated that the heightened expression of KIF23 mRNA was exclusive to TNBC cell lines. Furthermore, this higher expression was correlated with a worse prognosis and potentially facilitated the proliferation, migration, and invasion of TNBC both in laboratory settings and in living organisms. The advancement of EMT in TNBC was shown to be correlated with the activation of the Wnt/β-catenin pathway, as evidenced by western blot analysis. This observed mechanism was found to be connected to the aforementioned findings.56
On the other hand, Xin He et al. (2022) provided evidence that inhibiting the expression of KIF23 resulted in a decrease in the levels of phosphorylated glycogen synthetase kinase-3β (GSK-3β), β-catenin, cyclin D1, and c-myc in BC cells. This finding suggests that KIF23 less expression in BC contributes to the inactivation of the Wnt/β-catenin pathway, which reduces tumor growth. The researchers further confirmed this inhibitory effect by evaluating the impact of KIF23 downregulation in a xenograft assay, where protumoral effects mediated by KIF23 overexpression in BC were eliminated.54 In the same way, Wolter et al. (2017) demonstrated that silencing the expression of KIF23 and PRC1 (another hub gene identified in this study) strongly suppresses the proliferation of MDA-MB-231 cells, which are widely used for the in vitro experimental study of TNBC, indicating that both genes could be proposed as prognostic biomarkers and could be potential therapeutic targets for the therapy of BC.66 The last findings were reinforced by Wei Jian et al. (2021) by using siRNA against KIF23 on MDA-MB-231 cells, thus inhibiting their proliferation and migration and resulting in the suppression of EMT.67
Abnormal spindle microtubule assembly (ASPM), the other hub gene identified by all cytohubba algorithms, acts as a positive regulator of the Wnt/β-catenin signaling pathway, similar to KIF23.68 ASPM is a centrosomal protein that contributes to mitotic spindle formation, orientation, cytokines, neurogenesis, and brain development.69,70 During metaphase, ASPM is located at the spindle poles, whereas during interphase, it is found in the nucleus and centrosome. Moreover, the ASPM localization to the midbody during cytokinesis suggests that it participates in abscission.71 ASPM is also known to be involved in DNA repair, DNA replication, and cell cycle arrest.72
Typically, ASPM has been extensively associated with autosomal primary microcephaly, a brain disorder characterized by a normal-appearing but small brain and mental retardation.73 However, in the last decade, this gene has also been implicated in the progression of a variety of malignancies,71 including glioblastoma, cancers of the prostate, breast, bladder,74 and epithelial ovarian,71,73 as well as endometrial,69 hepatocellular,75 lung,76 and papillary renal cell carcinoma.70
ASMP has been recognized by several authors as a significant prognostic gene in cases of BC. The study of RNA sequencing (RNA-seq) data from 116 BC tissues lacking expression of ER, PR, and HER2, as well as 113 normal tissues obtained from the TCGA, provides more support for the aforementioned statement.77 On the other hand, Shubbar et al. (2013) identified this gene, along with CCNB2, CDCA7, KIAA0101, and SLC27A2 as molecular signatures in aggressive tumors associated with distinct clinical outcomes, based on their significantly dysregulated gene expression in relation to short-term disease-specific survival, triple-negative status, and/or histological grade stratification in 97 primary invasive diploid breast tumors.72
In their research, Xu et al. (2021) revealed that ASPM is necessary for DNA repair by homologous recombination (HR) and is functionally related to the BC susceptibility protein BRCA1. ASPM interacts with BRCA1 and HERC2 to prevent HERC2 from getting access to BRCA1 and to maintain its stability. Inhibiting ASPM expression increases HERC2-mediated BRCA1 degradation, impairs HR repair efficacy and chromosomal stability, and renders cancer cells more sensitive to ionizing radiation.78
Based on the above, it is plausible to propose that KIF23 and ASPM may exert a pro-tumorigenic role in BC via activating the Wnt/β-catenin pathway, postulating them as potential genes for the prognosis and diagnosis of this pathology.54,66,67,79
The other identified hub genes (AURKA, RACGAP1, CENPF, HMMR, and PRC1), as mentioned above, were identified using nine cytohuba algorithms; within these, PRC1 appears to play a central or connector role, ranking as the hub gene within the network. PRC1 exhibits the highest number of connections, suggesting its possible involvement in several signaling pathways.
Protein regulator of cytokinesis 1 (PRC1) belongs to the microtubule-associated protein superfamily and is involved in the regulation of cell proliferation and cytokinesis, the process by which a cell divides into two daughter cells at the end of the cell cycle. PRC1 facilitates the crosslinking of microtubules, thereby contributing to its essential role in these cellular processes. Additionally, it serves as a substrate for cyclin-dependent kinases and is intricately engaged in the interaction with microtubules and the creation of central spindles. The expression of PRC1 is notably seen during the G2 phase, hence exerting regulatory influence on spindle elongation and polarity. Therefore, the effective suppression of this process hinders the normal advancement of spindle elongation and polarization, leading to the cessation of the G2/M phase.80,81
The maintenance of genome integrity requires the proper execution of cytokinesis; failure in this process may lead to aneuploidy, a condition characterized by an abnormal number of chromosomes, which is known to contribute to the development of cancer. Previous studies have provided evidence supporting the existence of a correlation between PRC1 and chromosomal instability (CIN), a major characteristic of many cancers.81–83 A computer analysis showed that this gene exhibited the second highest position out of 10,151 genes examined, suggesting its association with a significant level of CIN across a wide range of tumor types. In addition, the inclusion of PRC1 within a CIN signature consisting of 25 genes was documented. The upregulation of these genes was shown to be correlated with heightened aneuploidy, leading to unfavorable prognoses for patients.84
On the other hand, it is well recognized that p53, Wnt/β-catenin, and the non-classical estrogen receptor pathways, which often undergo alterations or mutations in different types of malignancies, have the capacity to induce transcriptional dysregulation of the PRC1 gene.81 This dysregulation has been significantly linked with worse prognostic outcomes in several types of malignancies, including colon, liver, prostate, and BC.82,83
In a study conducted by Liang et al. (2019), PRC1 was able to increase the proliferation and migration rate of non-small cell lung cancer transfected with the high-expression PRC1 plasmid (TOPO-PRC1 group) by regulating p21/p27-pRB family molecules and the FAK-paxillin pathway. In the same way, these cells exerted a lower incidence of staying at the G2/M phase (P<0.05) than cells with the low-expression PRC1 plasmid.85
In the case of BC, several in vitro genomic studies have shown that PRC1 is among the genes that exhibit significant upregulation. This finding is further supported by western blot analysis, specifically during the G2/M phase. Notably, the administration of siRNA targeting PRC1 substantially lowered its expression and impeded the proliferation of BC cell lines such as T47D and HBC5.86 In the same way, 17-β-Estradiol (E2) can induce an upregulation of PRC1 expression in BC cells via a mechanism that is independent of the estrogen receptor (ER). This phenomenon is likely influenced by the formation of mitochondrial reactive oxygen species (mtROS); thus, antioxidants and mitochondrial blockers, which are unable to bind to the ER, may suppress the E2-mediated activation of PRC1. The specific method through which mtROS stimulates PRC1 expression remains unknown. However, the use of antioxidants and/or mitochondrial blockers might potentially be used to restore PRC1 levels in ER-negative BC.81,87
On the other hand, PRC1 is a transcriptional target of the Wnt/β-catenin pathway, and its subcellular location in the cytoskeleton is subject to dynamic regulation by Wnt3a protein. In contrast, it has been shown that PRC1 engages in interactions with the β-catenin destruction complex, therefore exerting regulatory control over the process of Wnt3a-induced membrane sequestration. This, in turn, facilitates the promotion of β-catenin signaling. Previously, we described the role of Wnt/β-catenin on BC and its modulation by other identified hub genes such as KIF23 and ASPM, thus demonstrating their interconnection around BC.81,88
Another gene associated with CIN, like PRC1, is CENPF, or the centromere protein F, responsible for encoding a protein that plays a crucial role in the process of chromosome segregation during cellular division.89 This protein has a remarkable degree of evolutionary conservation and is closely associated with the structure and function of the kinetochore, which is responsible for the organized pairing and segregation of chromosomes.90 The expression of CENPF is minimal during the G0/G1 phase but increases throughout the S phase, particularly in the nuclear matrix. Its greatest expression is seen during the G2/M phase.91,92
Similar to the aforementioned genes, CENPF has significant expression levels in several human tumor types, including pancreatic carcinoma, prostate cancer, and BC, where it has been recognized as a protein marker indicative of tumor cell proliferation.90,92 A variety of studies have provided evidence about the involvement of CENPF in the development, progression, and evolution of BC, hence supporting its candidacy as a pivotal gene for the diagnosis and prognosis of this disease.89–96
Sun et al. (2019) employed bioinformatic, computational, and western blotting methods to demonstrate that CENPF controls BC metastasis to bone through PI3K-AKT-mTORC1 signaling. This causes more secretion of PTHrP to be released and changes to the osseous environment of the host to encourage the formation of osteoclasts and the colonization of bone.91
In a study conducted in Sweden, the impact of single nucleotide polymorphisms (SNPs) in the CENPF gene on BC risk and clinical outcome was examined. The study included 749 incident BC patients with comprehensive clinical data and up to 15 years of follow-up, as well as 1493 matched controls. The findings revealed that individuals carrying the A allele of the CENPF SNP rs438034 had poorer BC-specific survival compared to those with the wild-type genotype GG (hazard ratio of 2.65, 95% confidence interval (CI) 1.19–5.90). However, these individuals were less likely to have regional lymph node metastases (odds ratio (OR) 0.71, 95% CI 0.51–1.01) and tumors of stage II–IV (OR 0.73, 95% CI 0.54–0.99).94
On the other hand, several bioinformatic and experimental studies have identified CENPF as a key gene with an important role in the development of different types of BC, including TNBC.90,93,95,96 All of them concluded that the overexpression of this gene is highly associated with a poor prognosis in patients, proliferation, and invasion. Among the mechanisms employed by CENPF on BC, it has been reported: i) DNA damage response and DNA repair through the control of the Rb-E2F1 axis (Chk1-mediated G2/M phase arrest by binding to Rb to compete with E2F1 transcription factor activity);90 ii) cell cycle regulation, DNA repair, Hedgehog pathway, histone phosphorylation;95 iii) PI3K/AKT/mTOR signaling.96
In addition to the aforementioned, hub genes such as HMMR and AURKA1 have been linked to BC through their interaction with tumor suppressor genes like BRCA1. HMMR (also known as RHAMM, XRHAMM, IHABP, and CD168) encodes the Hyaluronan Mediated Motility Receptor, whose functions include the regulation of stem cell proliferation via its ligand, hyaluronic acid (HA), an extracellular matrix polysaccharide essential for immunity, tissue differentiation. HA functions are linked to its molecular weight; thus, HA with a high molecular weight is responsible for structural functions, whereas HA with a low molecular weight interacts with cell receptors. The overproduction of HA with a high molecular weight is associated with arthritis, diabetes, and malignancy.97–99
HMMR is a coiled-coil protein that directly binds to microtubules through its N-terminus and finds its way to the centrosome through a bZip motif at its C-terminus; this motif is essential for interacting with TPX2 (an important component of the acentrosomal microtubule organizing center on neurons and Ran-dependent microtubule assembly near chromosomes), and Aurora kinase A activity; HMMR is also crucial for mitotic spindle orientation in human mitotic cells,97,98 chromosome segregation, and the assembly of the kinetochore and nuclear pore complex. The silencing or elevating of HMMR expression disrupts microtubule-based processes during cell division, resulting in mitotic spindle abnormalities, genome instability, and changes to the cell division axis.97,100 Due to its mitotic functions, proliferation-associated transcription factors (FOXM1, E2F, and MYC) and TP53 regulate the expression of the HMMR gene, which is co-expressed with TOP2A, KIF11, TPX2, BUB1, KIF20A, NUSAP1, KIF20B, SMC2, and CCNA. The silencing or upregulation of HMMR expression disrupts microtubule-based processes during cell division. HMMR is one of 14 proteins whose levels are highest during G2-phase and mitosis, along with other cell cycle gene products such as Aurora A and B, Polo-like kinase 1, and CENPF.97
HMMR, like other identified hub genes, has been linked to multiple tumor types, including acute myeloid leukemia, sarcoma, prostate cancer, lung cancer, and BC. Similarly, HMMR overexpression is strongly associated with bad pathological stages and poor patient survival rates.99 In BCs, the function of HMMR has been described at length. HMMR is expressed in breast tissue and forms a complex with BRCA1 and BRCA2.101 According to Xidong Ma et al. (2022), regulatory T cells (Treg) infiltrated by BC express this protein.100 In addition, Yeh et al. (2018) demonstrated that HMMR expression levels were considerably higher in IDC compared to luminal A and luminal B subtypes.102
HMMR is considered a low-penetrance BC susceptibility gene that interacts with BRCA1 to regulate cell division and apicobasal polarization of mammary epithelial cells and is a potential modifier of BRCA1-associated BC risk. HMMR acts as a key substrate for the BRCA1-BARD1 E3 ubiquitin ligase during spindle assembly. BRCA1 and HMMR interact to regulate microtubule structures involved in the correct apicobasal polarization of mammary epithelial cells; in fact, the dysfunction of BRCA1 stabilizes HMMR protein expression.97,101,103 Mateo et al. (2022) demonstrated that HMMR overexpression in mouse mammary epithelium increases BRCA1 mutant tumorigenesis by modulating cancer cell phenotype and tumor microenvironment via Aurora kinase A (AURKA) activation and decreasing Actin-related protein 2/3 localization 3 complex subunit 2 (ARPC2) in mitotic cells, which correlates with micronucleation and activation of cGAS-STING and non-canonical NF-B signaling. Initial tumorigenic events are characterized by genomic instability, EMT, and macrophage tissue infiltration.103
On the other hand, Aurora kinase A (AURKA) is an enzyme with serine/threonine kinase activity that regulates mitotic progression, centrosome formation, the establishment of the mitotic spindle throughout chromosome segregation during cell division, and the maintenance of chromosomal stability. AURKA is highly expressed at the end of the S phase in proliferating cells and accumulates until the G1 phase, or late mitosis, of the cell cycle.104–106 In a variety of cancers, AURKA functions as a key regulatory component in critical control points of response for cell oncogenic transformation by inhibiting the activity of p53/TP53 through the phosphorylation of the protein phosphatase 1 (PP1) isoforms.107 AURKA overexpression alters the sensitivity to microtubule drugs, resulting in chemotherapy resistance, according to studies conducted on various cancer cell lines. The abnormal expression and localization of AURKA regulate the occurrence and progression of tumors via multiple mechanisms; the primary pathways include accelerating tumor cell cycle progression, activating tumor cell survival or anti-apoptotic signaling pathways, inducing tumor cell genome instability, increasing tumor cell epithelial-mesenchymal transition, and promoting the formation of tumor stem cells with the capacity for self-renewal.108
Like other hub genes, the overexpression of AURKA has been detected in 96% of solid tumors (ovarian, colorectal, and BC tumors, as well as hematologic tumors such as T-cell lymphoma) and is associated with poor prognosis and drug resistance.109 AURKA is highly expressed in approximately 73% of BC tumors and functions as a mitosis regulator required for genome stability. It regulates the G2/M transition through the phosphorylation of BRCA1 and activation of the cyclin-dependent kinase 1 (CDK1)/cyclin B complex, thereby inhibiting apoptosis.105,106,108–110 Overexpression of AURKA and Bcl-xL induces EMT, analogous to KIF23 and HMMR, and is associated with BC metastasis.65,104,111 Several studies, including one conducted by Skov et al. (2022), demonstrated the aforementioned; these researchers identified gene amplification and protein upregulation of AURKA and Bcl-xL in basal B TNBC cell lines compared to basal A cells, which correlated with a mesenchymal phenotype and more invasive capabilities of basal B cells.109
On the other hand, nuclear localization of AURKA in BC has been linked to the transcriptional up-regulation and stabilization of oncogenes such as FOXM1 and myc family members; in fact, it has been shown that there is a positive feedback loop between AURKA and FOXM, which is essential for BC stem cell self-renewal.108,111,112 Finally, NF-B signaling is yet another pathway regulated by AURKA. The phosphorylation of IκBα by AURKA promotes its degradation, activating the NF-κB pathway, as revealed by a mechanistic study. YAP is the primary effector of the Hippo pathways downstream. The phosphorylation of YAP at Ser397 by AURKA is essential for the transcriptional activity and transformation mediated by YAP in TNBC.111
Finally, mitochondrial fragmentation, a phenomenon often seen in several experiments related to RACGAP1 (the last hub gene identified), has been identified as an additional signaling route linked with BC. The study conducted by Ran et al. (2021) provided evidence supporting the significant involvement of RACGAP1 overexpression in the metastasis of BC via the modulation of mitochondrial fragmentation, enhancement of mitophagy intensity, promotion of mitochondrial turnover, and augmentation of ATP generation through aerobic glycolysis. Moreover, the protein RACGAP1 was shown to facilitate the process of mitochondrial fission by binding ECT2 during the anaphase stage, which in turn triggers the activation of the ERK-DRP1 pathway. The overexpression of RACGAP1 also resulted in an increase in the expression of PGC-1a, a crucial regulator of mitochondrial biogenesis.113
The expression of RACGAP1 in human malignancies has been evaluated by several authors using various bioinformatics approaches. These studies have shown a significant association between RACGAP1 expression and worse prognosis in various human cancer types, including BC.114,115
The gene RACGAP1 has been identified as a potential oncogene in many types of human cancers. This is a component of the centralspindlin that is necessary for the activation of cytokinesis and looks to be a member of the Rho GTPase-activating protein family. The protein RACGAP1, when combined with GTP-bound Rac1, serves as a mediator for the tyrosine phosphorylation of the signal transducer and activator of transcription (STAT) protein family. Additionally, it acts as a nuclear chaperone for phosphorylated STATs, containing a nuclear localization signal. These phosphorylated STATs already possess various functions, such as antiapoptotic, proliferation, differentiation, and inflammation.116
In a two-arm study, 595 high-risk early BC patients were given surgical dose-dense progressive chemotherapy with epirubicin followed by cyclophosphamide, methotrexate and 5-fluorouracil with or without paclitaxel. The goal was to find out what role RACGAP1 mRNA expression plays in predicting disease-free survival (DFS) and overall survival (OS) in these patients. As a results high levels of RACGAP1 mRNA (above the median) were linked to lower DFS (log-rank, p = 0.002) and OS (p < 0.001). At the same way, a Cox multivariate regression analysis showed that high RACGAP1 mRNA expression independently predicted poor overall survival (p = 0.008). This means that high RACGAP1 mRNA expression is linked to a bad prognosis in early-stage BC patients with a high risk who are receiving dose-dense sequential chemotherapy.117
In conclusion, the use of integrated bioinformatics methodologies facilitated the discovery of seven important genes by examining three datasets obtained from the Gene Expression Omnibus (GEO). These genes, including KIF23, ASPM, AURKA, RACGAP1, CENPF, HMMR, and PRC1, have potential for predicting invasive ductal carcinoma (IDC) outcomes. To the best of our knowledge, there is currently no existing literature documenting these genes as biomarkers for invasive ductal carcinoma (IDC). However, they have been linked to other forms of BCs. This study contributes to the advancement of novel research endeavors focused on elucidating the mechanisms by which genes implicated in IDC exert their effects, using experimental models to provide empirical evidence and enhance understanding in this area.
Raw data derived from differential expression analysis performed in GEO2R for each IDC stage in the datasets:
Figshare: Raw data derived from differential expression analysis performed in GEO2R for each IDC stage in the datasets. https://doi.org/10.6084/m9.figshare.24310927.v1.118
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
Figsare: R script for identifying DEGs in datasets. Online resource. https://doi.org/10.6084/m9.figshare.24311218.v1.119
Figshare: Supplementary Figure S1: https://doi.org/10.6084/m9.figshare.24311200.v1.120
Figshare: Supplementary Figure S2: https://doi.org/10.6084/m9.figshare.24311209.v1.121
Figshare: Supplementary Figure S3: https://doi.org/10.6084/m9.figshare.24311188.v2.122
Figshare: Supplementary Figure S4: https://doi.org/10.6084/m9.figshare.24311251.v1.123
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)