Keywords
common ice plant, genome release, halophilism, salt-tolerance, salinity, photosynthesis
This article is included in the Bioinformatics gateway.
This article is included in the Plant Science gateway.
This article is included in the Genomics and Genetics gateway.
This article is included in the Japan Institutional Gateway gateway.
This article is included in the Bioinformatics Education and Training Collection collection.
common ice plant, genome release, halophilism, salt-tolerance, salinity, photosynthesis
We have removed six sentences, revised five groups of sentences, and enhanced the change by bold in the Discussion section. Also, we excluded the following six references which used to be listed in Reference section. In addition, our decoding genome sequence became publicly accessible on May 5th, 2023, at the National Center for Biological Information (NCBI) and the DNA Data Bank of Japan (DDBJ). Consequently, we have replaced the text in the 'Data availability' section under the 'Underlying data' section.
See the authors' detailed response to the review by Xin-Guang Zhu
See the authors' detailed response to the review by Qijie Guan
Soil salinity is one of the most detrimental abiotic stresses. Osmotic and ionic stresses can lead to decreased plant growth and economic damage, with estimates suggesting that it costs the global economy around $27.3 billion annually in lost crop yields (Qadir et al. 2014). Developing a wide range of strategies for adapting to and mitigating NaCl stress is required to address the negative impacts of salinity. Efficient resource management and crop improvement will help overcome the salinity-induced damages to agricultural production (Shrivastava and Kumar 2015). Mesembryanthemum crystallinum L. or the common ice plant is an annual plant of the family Aizoaceae, native to South Africa. This plant survives in the presence of a high salt concentration, even higher than that of seawater and can accelerate its growth under moderate salinity around 200 mM NaCl, wherein the growth and development of most crops are severely inhibited (halophilism; Agarie 2004). Also, it converts its photosynthetic mode from C3 to Crassulacean acid metabolism (CAM) under severe salt stress and drought stress (Adams et al. 1998). For the past half-century, the common ice plant has been frequently used as a model for elucidating the mechanisms of salt stress tolerance and photosynthetic conversion in response to salt and drought stresses.
Recently, the molecular processes underlying these phenomena have been elucidated at the levels of transcription, post-transcription (Taybi and Cushman 1999; Zhang et al. 2021), translation, post-translation (Forsthoefel et al. 1995; Nimmo 2000), specific proteins (tonoplast and plasma-membrane H(+)-ATPases: Vera-Estrella et al. 1999; glucose 6-phosphate/phosphate translocator: Kore-eda et al. 2013; ions transporter and compatible solute synthase: Tran et al. 2020a), mitochondria, and chloroplasts (Tran et al. 2020b; Niewiadomska and Pilarska 2021). Also, high-throughput gene expression profiling has been conducted using expression sequence tags (ESTs) (Kore-eda et al. 2004), microarrays (Cushman et al. 2008), and next generation sequencing (NGS; Oh et al. 2015; Tsukagoshi et al. 2015; Chiang et al. 2016; Kong et al. 2020).
These days, the comparative analysis of genome information focused on CAM-related genes in the common ice plants has been reported (Shen et al. 2022), but this genome resource is not easy to use. The poor transparency of genome information causes the delayed elucidation of whole-genome functions of the common ice plants dominating not only photosynthetic conversion systems but also halophilism and salt tolerance. The genomic sequences include protein-coding regions and untranslated regions such as promoters and terminators. MicroRNAs (miRNAs) and long noncoding RNAs (lncRNAs) influence gene expression through affecting mRNA stability and translation efficiency (Hughes 2006). Information regarding the genome sequences’ biological functions facilitates a comprehensive understanding of the transcriptional regulatory mechanisms of gene expression. Disclosure to researchers around the world is essential for clarifying the responsibilities of the entire genome to NaCl and creating superior cultivars through genome editing and selective breeding.
Short-read sequencing costs less than the long-read sequencing obtained using third and fourth NGS. Several software programs for de novo genome assembly for short reads have been developed. The algorithm for genome assembly (ALGA) is the newest assembler, based on an overlapping graphs model, which can generate more accurate results than conventional software using the de Bruijn graphs model (Swat et al. 2021). This achievement can be regarded as a model case of a genome study using NGS short reads, given its level of success.
In this study, we constructed the ice plant genome using easy-to-start applications such as ALGA to accelerate genome analysis. We investigated the characteristics of the genome, clarifying the repetitive sequences, tRNAs, and miRNAs (genomic regions and precursors), and identified gene regions using various software and web tools. This is the first report of whole-genome analysis of the common ice plant. Our results indicate the involvement of translated and untranslated regions in the regulatory processes of salt tolerance and photosynthetic conversion under stress in the ice plant.
All the processes involved in this study were archived in protocol.io (Sato et al. 2023a) and were described in Figure S8 (Sato et al. 2023b).
Seeds of the common ice plant (Mesembryanthemum crystallinum) were personally provided by Dr. John C. Cushman from the University of Nevada and stored under coolness and darkness until use. Originally, wild-type seeds were collected from the plants identified by Dr. Klaus Winter, an expert on the common ice plant, on a coastal cliff at the Mediterranean Sea shore close to Caesarea in Israel (around N32° 29′ 43.4″, E34° 53′ 22.8″) in 1978 (Winter et al. 1978). Three voucher specimens of M. crystallinum have been deposited in the Herbarium at the Royal Botanic Gardens Kew (55793.000, K000296094, and K000267571). In this study, our biological materials were recognized as the same plants as those specimens. Experiments, including collecting samples for this study, were conducted in compliance with relevant institutional, national, and international guidelines and laws. The seeds were aseptically sown on a medium for germination containing 4.6 g L-1 MS salt (mixed salts for Murashige-Skoog medium), 30 g L-1 sucrose, 1 mL-1 B5 vitamin (Gamborg et al. 1968), 1 g L-1 nicotinic acid, 1 g L-1 pyridoxine hydrochloride, 10 g L-1 thiamine hydrochlorides, and 100 g L-1 myo-inositol), 0.80% (w/w) agarose, and pH 5.7. The raising of seedlings was performed according to the methods published by Agarie et al. (2009). The two-week-old seedlings grown in a growth chamber under 12 h of light and 12 h of darkness at 25 °C were transferred to plastic pots filled with the growth medium soils composed of 50% peat moss, 30% cocopeat, and 20% perlite, tailored for the ice plants (Japan Agricultural Cooperatives Ito-Shima, Fukuoka, Japan). The plants were irrigated with a nutrient solution of 1.5 g L-1 OAT House No. 1, containing primary nutrients including 10% Nitrogen (1.5% as ammoniacal nitrogen and 8.2% as nitrate nitrogen), 8.0% water-soluble phosphoric acid, 27% water-soluble potassium, 4.0% water-soluble magnesium, 0.10% water-soluble manganese, 0.10% water-soluble boron, 0.18% iron, 2.0 × 10-3% copper, 6.0 × 10-3% zinc, and 2.0 × 10-3% molybdenum, in addition to 1.0 g L-1 No. 2, comprising 11% nitrogen and 23% lime (OAT Agrio Co., Ltd., Tokyo, Japan) in a greenhouse at Kyushu University for five weeks. The plants were treated with the solution including 51 mM NaCl for two weeks. Approximately 0.60 g of tissue from each leaf was collected, quickly frozen in liquid nitrogen, and stored at −80 °C.
Total genomic DNA was extracted from the leaf tissue and purified using MagExtractor™-Plant Genome Nucleic Acid Purification Kits (Toyobo Co., Ltd., Shiga, Japan), according to the manufacturer’s instructions. The DNA samples were fragmented by sonication and used to construct short insert paired-end libraries construction using NEBNext® Ultra™DNA Library Prep Kits for Illumina (New England Biolabs Ltd., Ipswich, MA, USA). Briefly, in the end-repair step, fragmented DNA was phosphorylated at the 5′ end and adenylated at the 3′ end. During the ligation step, full-length circulated adaptor sequences were ligated to the fragments. After adaptor cleavage, purification and size selection were performed. The indexed PCR products were taken to obtain the final sequencing libraries. The mean insert size for paired-end libraries was 300 bp. The paired-end (2 × 150 bp) sequencing was conducted on an Illumina NovaSeq 6000 platform (Illumina Inc., San Diego, CA, USA).
The mean insert size was calculated using REAPR (v1.0.18) (Hunt et al. 2013), and raw paired-end sequences were filtered based on the frequency of 21-mer sequences using the program Musket (v1.1) (Liu et al. 2013). The key parameter values were as follows: musket -omulti output -inorder pair1.fastq pair2.fastq. Sequence reads that appeared rarely or abnormally frequently were removed to obtain clean read data. In the corrected reads, unique and duplicate read numbers in the corrected reads were measured using fastqc (v0.11.9) (Simon 2010). The clean data were used for an estimate of genome size as follows. K-mers were counted and exported to histogram files using jellyfish (v2.3) (Marçais and Kingsford 2011) [key parameter: jellyfish histo reads.jf]. GenomeScope2.0 (Ranallo-Benavidez et al. 2020) corresponding key parameters were applied to calculate the genome sizes using k-mers lengths of 21 and 25.
The reads were assembled using ALGA (v1.0.3; Swat et al. 2021) with the default parameter --error-rate = 0.02. long DNA fragments 1 to 10 kb in length were combined, and gaps between them were filled with unknown bases (Ns) using Redundans (v0.14a; Pryszcz and Gabaldón 2016), a software program for scaffolding, with default parameter values. The genome coverage of reads was estimated using Mosdepth program (Pedersen and Quinlan 2018). The completeness of the assembled genome was evaluated based on the content of orthologs in higher plants, using the benchmarking universal single-copy orthologs (BUSCO) program (v5.0; Manni et al. 2021). The lineage dataset was embryophyta_odb10 (creation date: 2020-09-10, number of BUSCOs: 1614). We also searched for core genes in the genome sequences of nine other plant species: Kewa caespitosa, Pharnaceum exiguum, Macarthuria australis, Solanum chaucha, Populus trichocarpa, Arabidopsis thaliana, and Oryza sativa using BUSCO. The first three species belong to the same order, Caryophyllales, to which the ice plants belong. Genome information was obtained from the NCBI (see Note 1 “Address to genome information”, Sato et al. 2023b). The number of bases, sequences, sequences in several base number ranges, and maximum base length of the final draft genome sequences was calculated using gVolante (v2.0.0) (Nishimura et al. 2017). BLASTN (v2.2.31+; McGinnis and Madden 2004) was used to investigate the number of cDNA sequences identified by transcriptome (Lim et al. 2019), and registered DNA sequences (retrieved from NCBI, last accessed February 2022) were aligned to the final assembled genome sequence.
The 18S ribosomal genes were extracted using barrnap (v0.9; Seemann 2018) from the obtained genome sequences of the ice plant. As comparative objectives, 25 kinds of 18S ribosomal genes from general crops (Japanese radish [Raphanus sativus], Soybean [Glycine max], Japanese trefoil [Lotus japonicus], Barrelclover [Medicago truncatula], Adzuki bean [Vigna angularis], Banana [Musa acuminata], Barley [Hordeum vulgare], Sorghum [Sorghum bicolor], Bread wheat [Triticum aestivum], Maize [Zea mays], Apple [Malus domestica], Peach [Prunus persica], Coffee tree (Arabica var.) [Coffea arabica], Coffee tree (Robusta var.) [C. canephora], Clementine [Citrus clementina], Orange [C. sinensis], Poplar, Tobacco [Nicotiana tabacum], Tomato [Solanum lycopersicum], Eggplant [S. melongena], Potato [S. tuberosum] and Grape [Vitis vinifera]) were selected using the SILVA database 138.1 (Release. 2020-08; Pruesse et al. 2007). After joining all ribosomal DNA sequences into one file, a molecular phylogenetic tree was created using implemented in NGPhylogeny.fr (Lemoine et al. 2019) (Released in 2019). SH-aLRT (Shimodaira-Hasegawa-approximate likelihood ratio test) (Shimodaira and Hasegawa 1999) was used to determine the molecular phylogenetic tree.
Repetitive sequences were detected, and custom repeat libraries involving transposable elements and long terminal repeat-retro transposons were generated using RepeatModeler2 (v2.0.2; Flynn et al. 2020) and TEclass (v2.1.3; Abrusán et al. 2009). Known repeat sequences were detected and classified in the assembled genome sequence with reference to the Repbase library (Bao et al. 2015) and the custom repeat libraries, using RepeatMasker (v4.1.2-p1; Smit et al. 2013-2015). The capital letters in the genome sequences were replaced with small characters as soft masking.
The tRNA genes were identified in the draft common ice plant genome using tRNAscan-SE2.0 (v2.0.9) (Chan et al. 2021). The tRNA data of other nine plant species—Arabidopsis, rice, tomato, poplar, horseradish, potato, grape, soybean, and coffee tree (robusta species)—were obtained from the PlantRNA database (Cognat et al. 2013). The percentages of arbitrary tRNAs against the total tRNAs in the genome were calculated and compared to the ice plants’ values with those of the other species. Smirnov-Grubbs’ outlier tests were performed to select tRNAs more significantly involved. The test statistic T was calculated using the following equation:
The miRNA loci in the genome sequence were identified using the cmscan command in Infernal (v1.1.4; Nawrocki and Eddy 2013) using Rfam.
The BRAKER2 pipeline (v2.1.5; Brůna et al. 2021) was used for the prediction of genes in the common ice plant genome. Amino acid sequences were translated from the transcriptome profile reported by Lim et al. (2019) and used as additional reference data for the prediction of genes. BRAKER2 was used with the default parameters (–softmasking). The total sequences, total bases, total amino acids, and N50 were computed based on the resulting fasta-format files containing information about the genes, coding sequences, and amino acids using seqkit (v2.0.0; Shen et al. 2016) [key parameter: seqkit stats]. Protein BLAST searches (E-value < 1e-5) were conducted using DIAMOND (v2.0.13.151; Buchfink et al. 2021) against the NCBI-non-redundant protein sequences (retrieved from NCBI in March 2022), Uniprot-swissprot (retrieved in March 18), Ensemble TAIR10 (retrieved in March 2022), and NCBI poplar amino acid sequence databases (retrieved from NCBI in March 2022).
The protein domains in the genome were identified using the Pfam (v33.1) database (Mistry et al. 2021) with E-value < 1e-3, using HMMER (v3.1b2; Potter et al. 2018). The protein databases of rice, maize, and poplar from the NCBI (last accessed February 2022) were used in the domain for a detailed classification of the PKinase family, the iTAK (v18.12) web tool (Zheng et al. 2016; last accessed February 2022) was utilized. The ratio of families with a high ratio of genes to total genes in the ice plant was compared with that of the same families in the other plants. For statistical analysis, we used Smirnov-Grubbs’ outlier tests. The following equation was used to obtain the test statistic T:
Finally, BLASTP was used to compare proteins generated from the ice plant genome and those from Arabidopsis, rice, maize, and poplar and renamed TAIR10 ID. These IDs were subjected to gene ontology (GO) enrichment analysis using DAVID (updated in 2022; accessed on March 24; Sherman et al. 2022) based on a modified Fisher exact probability test with E-value < 0.05.
Short insert reads data (300 bp; Figure S1-(A), Sato et al. 2023b) with an estimated coverage of 50.92× and a ratio of unique to duplicate reads of about 1.63:1 was obtained by removing erroneous reads of raw paired-end data from the Illumina platform (BioProject: PRJDB13817; BioSample: SAMD00508673) (Table S1). The M. crystallinum genome size was estimated to be 366 to 369 Mb, with very low heterozygosity (about 0.010%) following an analysis of the frequency of 21 and 25-mers, using GenomeScope2.0 (Figure S1-(B) and (C), Sato et al. 2023b). The M. crystallinum final draft assembly included 286 Mb in 49,782 scaffolds with a scaffold N50 of 10,562 bp (Table S2). The BUSCO tool revealed 1,509 (93.49%) of 1,614 embryophyte library core genes, with 1,223 (75.77%) of these being ‘Complete’ matches in the genome. The completeness and contiguity of the genome were greater than the shotgun assembled M. australis and S. chaucha genomes (Figure S2: Sato et al. 2023b). Around 24,081 (99.5%) of the 24,204 transcripts from the transcriptome assembly of M. crystallinum leaves (Lim et al. 2019), and all 135 DNA sequences registered in the NCBI, were aligned to the assembled genome (Supplementary Dataset S1: Sato et al. 2023c).
We performed a phylogenic analysis using 18S ribosomal DNA (rDNA) among related species. The seven types of 18S rDNA were chosen from the ice plant genome sequence. Also, the other 25 plant species’ 18S rDNA sequences (see Phylogenetic tree creation among multiple plant species using 18S ribosomal DNA sequences in Methods) were retrieved from the ribosomal RNA database in SILVA and were aggregated with the ice plant 18S rDNAs. Based on these sequences, a molecular phylogenetic tree of 18S rDNA was constructed using PhyML+SMS/One Click in NGPhylogeny.fr (Figure S3, Sato et al. 2023b). The results showed that five species of 18S rDNAs were relatively closely related to poplar’s 18S rDNA.
In the 286.0 Mb M. crystallinum genome, 2,423 distinct repetitive sequences families, accounting for 110.9 Mb (38.8%) of the genome, were identified using custom repeat libraries and Repbase (Bao et al. 2015). This ratio was smaller than Shen et al.’s (2022) reported value (48.04%). In decreasing order of frequency, the annotated repetitive elements were unclassified 78.0 Mb (27.27%), retroelements 21.9 Mb (7.64%), long interspersed nuclear elements (LINE) 12.5 Mb (4.37%), long terminal repeats (LTR) 9.35 Mb (3.27%), and simple repeats 7.26 Mb (2.54%). Some retroelements were classified into subfamilies, including L1/CIN4 12.4 Mb (4.34%) and RTE/Bov-B 0.85 Mb (0.03%) in the LINE, and Ty1/Copia 4.90 Mb (1.71%) and Gypsy/DIRS1 4.35 Mb (1.52%) in the LTR (Table 1).
| Group | Number of elements | Length occupied, [bp] | Percentage of sequence, [%] |
|---|---|---|---|
| Retroelements(1) | 42,672 | 21,857,207 | 7.64 |
| LINEs(2): | 25,415 | 12,509,921 | 4.37 |
| RTE/Bov-B | 278 | 84,896 | 0.03 |
| L1/CIN4 | 25,137 | 12,425,025 | 4.34 |
| LTR elements(3): | 17,257 | 9,347,286 | 3.27 |
| Ty1/Copia | 9,514 | 4,904,145 | 1.71 |
| Gypsy/DIRS1 | 7,528 | 4,345,997 | 1.52 |
| DNA transposons(4) | 5,725 | 2,789,199 | 0.98 |
| hobo-Activator | 728 | 285,258 | 0.10 |
| Tc1-IS630-Pogo | 237 | 160,874 | 0.06 |
| Tourist/Harbinger | 590 | 263,259 | 0.09 |
| Rolling circles | 121 | 112,702 | 0.04 |
| Unclassified: | 392,582 | 77,986,137 | 27.27 |
| Total interspersed repeats: | 102,632,543 | 35.88 | |
| Simple repeats: | 137,610 | 7,255,343 | 2.54 |
| Low complexity: | 19,059 | 911,926 | 0.32 |
A total of 971 tRNAs, excluding pseudogenes, were detected in the assembled genome, and were sorted into several groups based on codon designation. The codon with the most abundant tRNA was isoleucine and the least was tryptophan (Figure S4, Sato et al. 2023b). The number of tRNAs was as follows: Arabidopsis 585, rice 505, poplar 505, tomato 723, horseradish 500, potato 736, grape 391, and soybean 700. Interspecific comparisons using the Smirnov-Grubbs outlier test and focusing on these eight species indicated that the abundance of isoleucine was significantly highest and that of tryptophan was significantly lower (P < 0.05; Figure 1 and Table S3, Sato et al. 2023b).
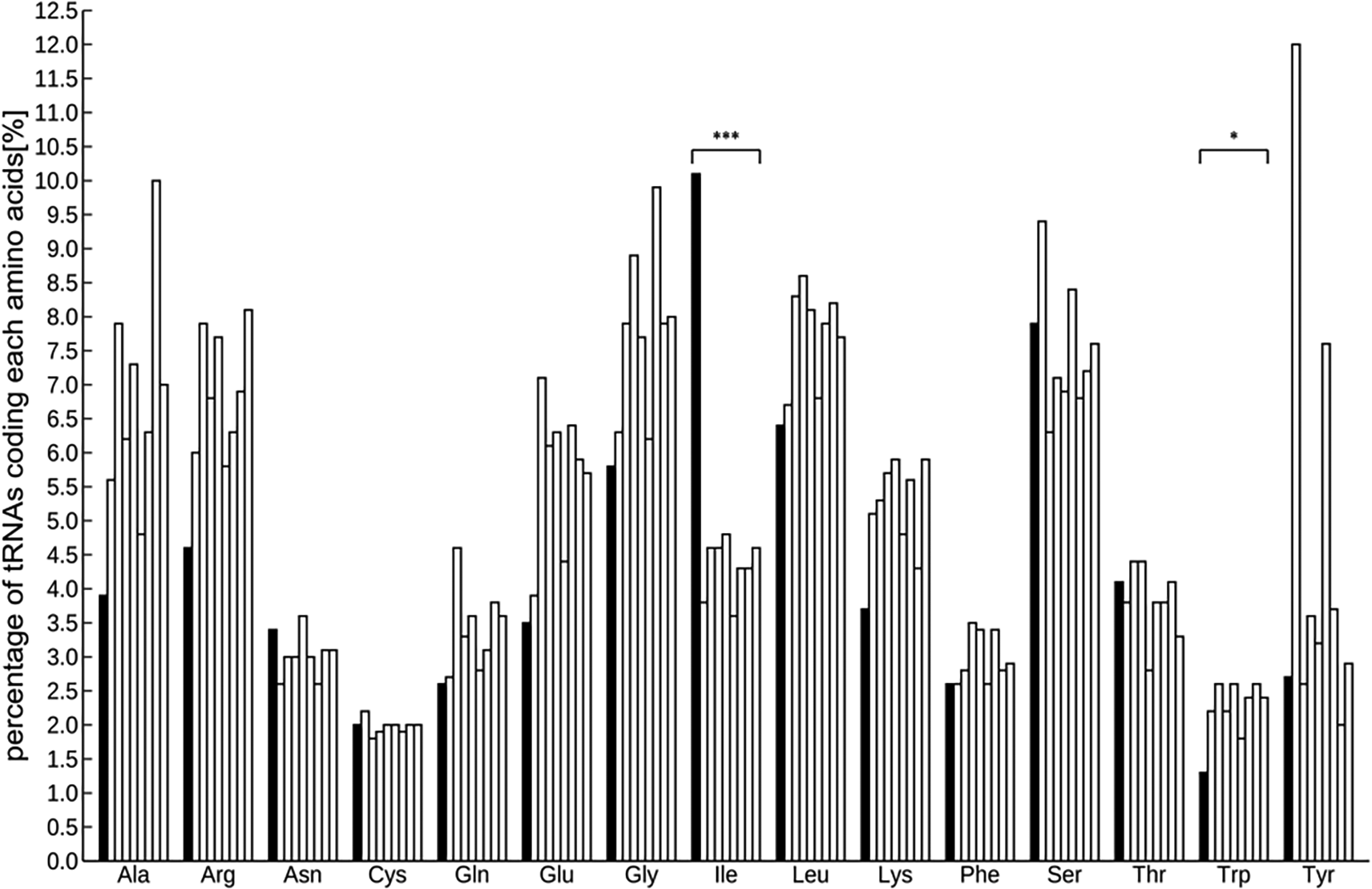
tRNAs significant differently abundant from the other 8 species by Smirnov-Grabs outlier test, are shown in black, and the other tRNAs are shown in gray. Bars indicate ice plant, Arabidopsis, rice, tomato, poplar, horseradish, potato, grape, and soybean from the left of each series. Asterisks indicate statistical significance: * P < 0.05, n = 9.
In addition, miRNAs loci were identified from the genome with reference to the Rfam database, to obtain miRNA profiling independent of their expression levels. MiRNAs are 21 to 24 nt molecules that regulate post-transcriptional mRNA modification, playing important roles in plant growth and tolerance to environmental stress. 100 miRNA loci were identified and categorized into 25 families. The RNA family with the largest number of loci was MIR169 (25), followed by mir-399 (16), MIR159 (8), and mir-166 (7). mRNAs targeted by miRNA families were predicted (Table S4). For instance, MIR169 family miRNAs were presumed to bind to mRNAs encoding nuclear factor gamma subunit A (NF-YA) (Chiang et al. 2016). Overall, 13 types of 25 miRNA families were likely to target mRNAs encoding transcription factors: MYB33, MYB65, HD-ZIP, WRKY, AP2-like, NAC, ARFs, IAR3, ARF16, OsSPL14, SPL, GRF2, and HLH. The rest of the targeted mRNAs are anticipated to have functions in processes such as miRNA maturation, mRNA cleavage, or metal binding.
Genes (34,223), coding sequences (35,702), and amino acid regions (35,702) were predicted from the soft-masked draft M. crystallinum scaffolds ab initio using a homology-based pipeline in BRAKER2 using transcriptome data (Table S4). The representative value on bases showed that coding sequence regions cover at least 10.6% (30.4 Mb) of the total genome sequence. In comparison to several databases on 25 plant species’ genes registered in PGDBj (Asamizu et al. 2014; last accessed in March 2022), the ice plants’ genes were as abundant as those of Sorghum bicolor and Arabidopsis lyrate. Additionally, summarized data indicated that the M. crystallinum gene number was 16 times larger than those of S. bicolor and A. lyrate, equivalent to about 27.6% of the number of genes of Triticum aestivum (bread wheat) and 3.31-fold greater than that of Pyropia yezoensis (bangia) (Figure S5, Sato et al. 2023b). Each translated protein sequence was used in a BLASTP search with the DIAMOND program (Buchfink et al. 2021) against four kinds of protein sequence databases. In order of the proportion of homologous amino acid sequences identified, they were NCBI-non-redundant (82.59%), poplar (70.65%), TAIR10 (65.39%), and Swiss-prot (56.05%; Table 2) (Supplementary Dataset S2: Sato et al. 2023d). To simplify gene ID conversion to GO terms, the results, including TAIR ID, were used in the functional estimation.
A Pfam domain search based on the Pfam (Mistry et al. 2021) database identified 3,703 domains in 23,521 (97.1%) genes. The most frequently occurring domain was the protein kinase domain (PKinase), at 2.18%, followed by a domain of unknown function (DUF) 4238 (1.85%), reverse transcriptase (RVT)_1 (1.45%), PPR domain-containing protein (PPR)_2 (1.42%), and protein tyrosine and serine/threonine kinase (PK_Tyr_Ser-Thr) (1.18%) (Supplementary Dataset S3: Sato et al. 2023e). The PKinase family was further classified into 94 kinase families using iTAK (Zheng et al. 2016). The top 30 kinase families with the largest number of ice plant genes are shown in descending order in Figure 2. Compared to the other four plant species, the proportion of 12 families was significantly higher (P < 0.05), and eight families—DUF4238, RVT_1, RVT_2, RVT_3, Retrotrans_gag_2, zf_RVT, Retrotrans_gag_3, Retrotrans_gag—contained retroelement domains that could be attributed to a retrotransposable element (Figure 3). The annotated genes were assigned to GO classifications based on TAIR ID in three groups —biological process (BP), cellular component (CC), and molecular function (MF)—and were categorized into 403 GO terms using the gene functional classification tool in the DAVID web service. The proportion of genes assigned to 94 GO terms did not differ significantly among five plant species (P > 0.05; Figure S6 to S8, Sato et al. 2023b), indicating that they are essential to plant survival. These findings confirmed that the ice plant genome constructed in this study contained conserved genes to some extent. 18 GO terms were identified only from ice plants, although the number of genes was small (Table S6), involving virus resistance, pollen tube development, and fat biosynthesis (BP); cytoplasmic vesicle and “soluble NSF attachment protein receptor” (SNARE; CC); and O-acyltransferase for transferring fatty acids (MF).
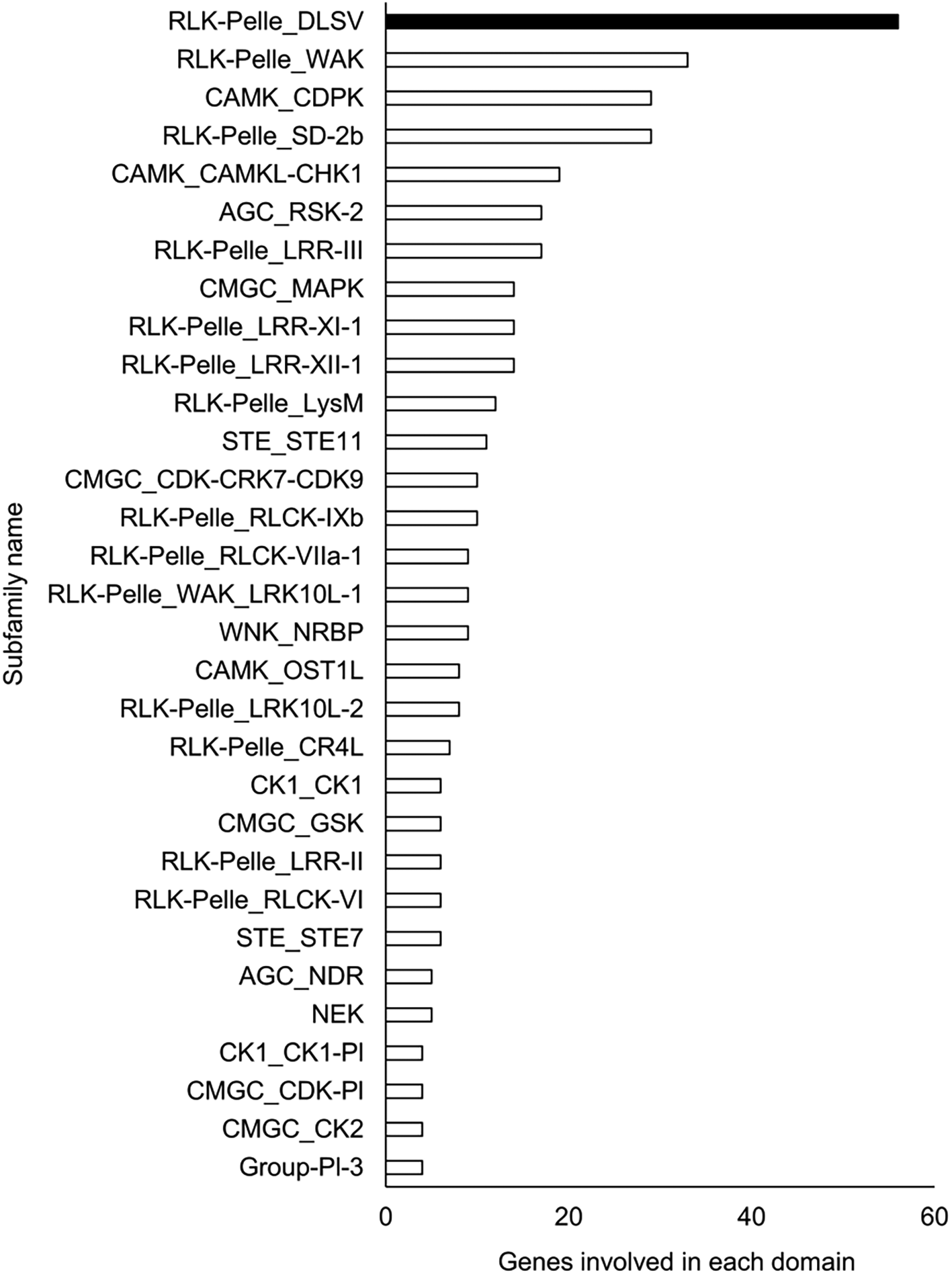
The family with the highest number of genes is shown in black, and the other families are shown in white.
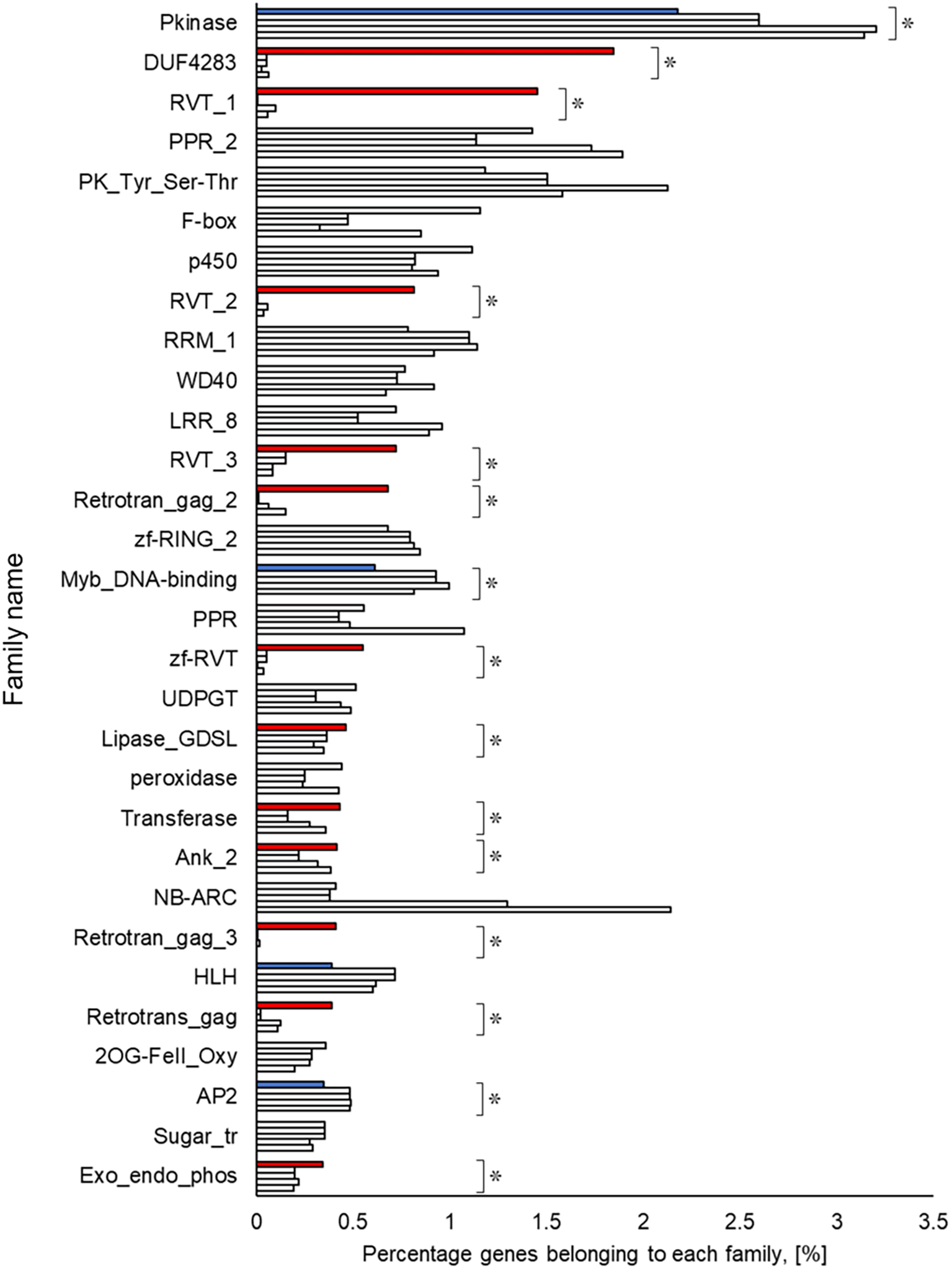
The top row for each family shows ice plant, Arabidopsis, rice, maize, and poplar. The independence of the proportion of genes belonging to a family in the ice plant is displayed using the Smirnov-Grubbs rejection test. Asterisks (*) indicate statistical significance: P < 0.05, n = 5. Independence is shown in red if the proportion is independently high in ice plant, in blue if it is low, and in gray if there is no difference.
M. crystallinum is utilized as a model plant for investigating halophilism, salt tolerance, and CAM photosynthesis. In this research, we have assembled the common ice plant’s genome sequence and elucidated the genome’s function in detail for the first time in this species. This genomic resource covers all protein-coding, non-transcribed, and untranslated regions. Our results of genomic functional analysis on M. crystallinum provide new clue to solve the molecular mechanisms underlying the ice plant’s adaptation to NaCl stress including conversion of the photosynthesis.
The total assembled genome length (286 Mb) was approximately 26% smaller than the genome size estimated using the experimental or bioinformatic method reported by Meyer et al. (1990; 390 Mb), de Rocher et al. (1990; 390 Mb), and Shen et al. (2022; 378 Mb). The genome size estimated using k-mer distribution analysis is likely to be smaller than that using experimental data, including flow cytometry, given the effects of repetitive sequences and other obscure nucleotide sequences (Bennett et al. 2003; Al-Qurainy et al. 2021). Barkla et al. (2018) have shown that the ploidy levels of the leaf increased throughout its development. Because polyploidy is becoming a concern when NGS is used for genome assembly (Kyriakidou et al. 2018), the endopolyploidy of ice plant leaves may increase the complexity of genome assembly. Experimental data is supposed to help to support the present results and determine the exact ice plant genome size.
Interestingly, the phylogenic tree analysis suggested that the genome composition of the ice plant was similar to that of the poplar. Previous studies have reported poplar-derived genes for salt tolerance, including PtNF-YA9 (Lian et al. 2018), PtSAP13 (Li et al. 2019), and PtVP1.1 (Yang et al. 2015). These results suggest that the ice plant genome constructed in this study is a highly conserved sequence that can be used for phylogenetic relationship analysis.
We found that the repetitive M. crystallinum sequences occupy 110.9 Mb (38.8%) of the genome. Advances in genomics over several decades have revealed that repetitive sequences play essential roles in regulating gene expression in higher plants. Recent studies observed that the transposable elements, involving many repetitive sequences were highly expressed under heat, salt, and intense light stresses, in Arabidopsis, tomato, and mangrove species (Deneweth et al. 2022; Wang et al. 2022). Further studies indicated that cis-regulatory motifs associated with C4 photosynthesis, rate-determined by the same enzyme up to at least 669, and the non-coding RNAs regulating methyltransferases expression levels are derived from transposable elements (Nosaka et al. 2012; Cao et al. 2016). Transposable element expression is suppressed by cytosine methylation in DNA sequences, chromatin remodeling, and degradation by small interfering RNA (siRNA; Ito 2013). These previous results suggested that the common ice plant has repetitive sequences with similar effects on gene expression regulation.
Two kinds of representative small non-coding RNAs were found in the ice plant genome—971 tRNAs and 100 miRNA loci—which are anticipated to be relevant to metabolic pathway and post-transcriptional modification. Generally, a tRNA recruits an amino acid corresponding to its codon, which means that the abundance of a specific tRNA is proportional to that of the relevant amino acid. Some studies have shown the effectiveness of amino acids in metabolism for environmental stress reduction. The Smirnov-Grabs outlier test indicated a notable abundance of tRNAs coding for isoleucine and a scarcity for tryptophan, pointing to a unique amino acid distribution in the ice plant compared to eight other plants, potentially contributing to its stress tolerance mechanisms.
Some miRNAs identified in the ice plant’s genome appeared to be key small molecules in the stability of mRNAs coding for epigenetic and transcription-related factors. The mRNA coding NF-YA were targeted by 31 MIR169 loci known to integrally regulate gene expression by maintaining histone acetylation in soybeans (Lu et al. 2021), or binding to circadian rhythm-related elements, including the “CCAAT” motif in Arabidopsis (Wenkel et al. 2006; Zhao et al. 2016). Several miRNA-targeting transcription factors were associated with salt tolerance (HLH, SPL, and HD-ZIP) (Shen et al. 2019; Wang et al. 2019, 2021) or CAM photosynthesis (WRKY, AP2, MYB, and NAC) (Amin et al. 2019; Yuan et al. 2020; Shah et al. 2021). All target gene families were found in the protein family collection in the ice plant genome, except for SPL and lectin receptor kinase (see Supplementary Dataset S1, Sato et al. 2023c), indicating that an antagonistic relationship between miRNAs and mRNAs underlies the stress tolerance and photosynthetic conversion mechanisms of the ice plants. Additional miRNA sequence information is expected to provide more accurate data and form the basis for testing these assumptions.
The richest PKinase subfamily was “receptor-like kinase/Pelle, DUF26, SD-1, LRR-VIII and VWA, a moss-specific new RLK subfamily (RLK-Pelle_DLSV)”, containing primarily receptor-type kinases, which was consistent with the transcriptome profiling in a halophyte, Nitraria sibrica (Zhang et al. 2022). It has been assumed to be involved in cell wall biosynthesis, adhesion, and developmental regulation. The common ice plants show halophilism or salt tolerance; a detailed study may help to shed light on the mechanism of this tolerance from the perspective of phosphorylation. In contrast to the rare PKinase, the richness of retrotransposon-derived domains (reverse transcriptase and gag genes), involved in RNA packaging and the replication cycle (Orozco-Arias et al. 2019), was apparent in the ice plant compared to the other plant species. Lipases, transferases, and phosphatases were abundant, and transcription factors such as Myb, HLH, and AP2 were scarce in the genome of the common ice plant. Two reviews show these enzymes and transcription factors assume a key role in plants’ survival under salinity (Reyes-Pérez et al. 2019; Chaudhry et al. 2021), then the elucidation of these protein interactions by transcriptome and interactome analysis may provide crucial evidence about their unknown functions.
Finally, comparing the gene functions among the genomes of five plant species—the common ice plant, Arabidopsis, rice, maize, and poplar—based on their gene counts, 18 gene functions were found only in the ice plant. Previous studies (12 reviews and 11 research articles) with sophisticated experimental backgrounds indicated that all gene functions were possibly associated with the mechanisms of halophilism, salt tolerance, and photosynthetic conversion. These gene functions were categorized as related to biological defense, growth, reproduction, transcription, post-transcription, and intermembrane transportation. Therefore, focusing on the homologous of the ice plant genes with these functions may provide critical insight into the salt-induced growth and photosynthetic systems.
We succeeded in assembling the M. crystallinum genome using Illumina PE reads, characterizing the genome, and identifying the potential gene, non-transcriptional and translational regions, and repetitive sequences. Furthermore, we made the ice plant genome available to all, which means the end of this plant’s genome information opacity temporarily. Our results revealed that salt tolerance increases with growth, and C3-CAM photosynthetic conversion in the presence of NaCl is probably controlled by both protein-coding genes and potential genomic factors, including transposable elements, tRNAs, miRNAs, and protein kinases. These findings provide new insights into the mechanisms of plant growth under environmental stresses and can be used to develop highly high salt-tolerant crops. We hope this study will be a good step stone to the developed genomic science of the common ice plant.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)