Characterizing alpha helical properties of Ebola viral proteins as potential targets for inhibition of alpha-helix mediated protein-protein interactions
[version 2; peer review: 1 approved]
Sandeep Chakraborty1,2, Basuthkar J. Rao2, Bjarni Asgeirsson3, Abhaya M. Dandekar1
Sandeep Chakraborty1,2, Basuthkar J. Rao2, Bjarni Asgeirsson3, Abhaya M. Dandekar1
1Plant Sciences Department, University of California, Davis, 95616, USA 2Department of Biological Sciences, Tata Institute of Fundamental Research, Homi Bhabha Road, Mumbai, 400 005, India 3Science Institute, Department of Biochemistry, University of Iceland, Dunhaga 3, IS-107 Reykjavik, Iceland
This article is included in the Ebola Virus collection.
Abstract
Ebola, considered till recently as a rare and endemic disease, has dramatically transformed into a potentially global humanitarian crisis. The genome of Ebola, a member of the Filoviridae family, encodes seven proteins. Based on the recently implemented software (PAGAL) for analyzing the hydrophobicity and amphipathicity properties of alpha helices (AH) in proteins, we characterize the helices in the Ebola proteome. We demonstrate that AHs with characteristically unique features are involved in critical interactions with the host proteins. For example, the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain has an AH with a large hydrophobic moment. The neutralizing antibody (KZ52) derived from a human survivor of the 1995 Kikwit outbreak recognizes a protein epitope on this AH, emphasizing the critical nature of this secondary structure in the virulence of the Ebola virus. Our method ensures a comprehensive list of such `hotspots'. These helices probably are or can be the target of molecules designed to inhibit AH mediated protein-protein interactions. Further, by comparing the AHs in proteins of the related Marburg viruses, we are able to elicit subtle changes in the proteins that might render them ineffective to previously successful drugs. Such differences are difficult to identify by a simple sequence or structural alignment. Thus, analyzing AHs in the small Ebola proteome can aid rational design aimed at countering the `largest Ebola epidemic, affecting multiple countries in West Africa' (http://www.cdc.gov/vhf/ebola/outbreaks/2014-west-africa/index.html).
Corresponding author:
Sandeep Chakraborty
Competing interests:
No competing interests were disclosed.
Grant information:
AMD wishes to acknowledge grant support from the California Department of Food and Agriculture PD/GWSS Board. BJ acknowledges financial support from Tata Institute of Fundamental Research (Department of Atomic Energy). Additionally, BJR is thankful to the Department of Science and Technology for the JC Bose Award Grant. BA acknowledges financial support from the Science Institute of the University of Iceland.
The revised version of the manuscript incorporates changes suggested by one reviewer. Specifically, we have clarified that the neutralizing antibody (KZ52) does not disrupt the GP2 structure,but binds to an epitope that prevents it from refolding, and abrogates its fusion in the host membrane. Also, we have modified the Methods sections to explicitly state that the proportion of charged residues is computed based on the total number of charged residues, and not the length of the helix. Lastly, we cite new research that uses computational methods (DOCLASP, PAGAL) to correlate the different immunosuppressive and pathogenicity mechanisms in Ebola and Marburg viruses to variations in their structures/sequences.
The revised version of the manuscript incorporates changes suggested by one reviewer. Specifically, we have clarified that the neutralizing antibody (KZ52) does not disrupt the GP2 structure,but binds to an epitope that prevents it from refolding, and abrogates its fusion in the host membrane. Also, we have modified the Methods sections to explicitly state that the proportion of charged residues is computed based on the total number of charged residues, and not the length of the helix. Lastly, we cite new research that uses computational methods (DOCLASP, PAGAL) to correlate the different immunosuppressive and pathogenicity mechanisms in Ebola and Marburg viruses to variations in their structures/sequences.
The Ebola virus was first discovered in 19761, and has been since known as a rare, but deadly disease2. However, the current outbreak in West African countries (Guinea, Liberia, Nigeria, Sierra Leone and Senegal) has rapidly deteriorated into a full blown epidemic3, and poses grave humanitarian dangers to these countries4. Ebola, along with the Marburg virus, belongs to the Filoviridae family5, and causes haemorrhagic fever2 by quickly suppressing innate antiviral immune responses to facilitate uncontrolled viral replication6.
Interestingly, the genome of the Ebola virus encodes seven proteins7, although their extreme ‘plasticity allows multiple functions’8,9. Protein structures are formed by well ordered local segments, of which the most prevalent are alpha helices (AH) and β sheets. AHs are right-handed spiral conformations which have a hydrogen bond between the carbonyl oxygen (C=O) of each residue and the alpha-amino nitrogen (N-H) of the fourth residue away from the N-terminal. AH domains are often the target of peptides designed to inhibit viral infections10–12. Recently, we have provided open access to software that has reproduced previously described computational methods13 to compute the hydrophobic moment of AHs (PAGAL14).
In the current work, we characterize the helices in the Ebola proteome using PAGAL, and demonstrate that the helices with characteristically unique feature values are involved in critical interactions with the host proteins. The PDB database is queried for the keyword ‘Ebola’, and the structures obtained are analyzed using DSSP (Define Secondary Structure of Proteins)15 for identifying AHs. We process all PDB structures, and do not filter out redundant structures based on sequence. These helices are analyzed using PAGAL, and the results are sorted based on three criteria - hydrophobic moment and high proportion of positive or negative residues. The helices that are ranked highest in these sorting criteria are involved in critical interactions with either antibodies or host proteins. For example, the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain has an AH with the largest hydrophobic moment in all helices analyzed16. This helix has part of the epitope recognized by the neutralizing antibody (KZ52) derived from a human survivor of the 1995 Kikwit outbreak, emphasizing the critical nature of this helix in the virulence of Ebola17. Another example, obtained by choosing the helix with the highest proportion of negatively charged residues, is the interaction between the human karyopherin alpha nuclear transporters C terminus and the Ebola virus VP24 protein (eVP24)18, which suppresses tyrosine-phosphorylated STAT1 nuclear import19. These helices probably are, or can be, the target of molecules designed to inhibit AH mediated protein-protein interactions20. Our method provides a comprehensive list of such targets. Further, each protein can be individually queried using PAGAL, and thus identified helices that might have a poor global rank, but still be critical in the particular proteins context.
Although, Ebola and Marburg viruses are members of the Filoviridae family21, they have different antigenicity of the virion glycoprotein22. By comparing the AHs in proteins of Marburg and Ebola viruses, we are able to elicit subtle changes in the proteins that might render them ineffective against previously successful drugs. These differences are not apparent from a simple sequence or structural alignment. Thus, in the current work, we elucidate a simple methodology that can aid rational design of drugs and vaccine, an important aspect of the global effort to counter the deadly Ebola epidemic.
Materials and methods
We searched for the keyword ‘Ebola’ in the PDB database (Table 1). Subsequently, each protein was split based on the chain ID, resulting in 146 single chained proteins (See ALPHA.zip in Dataset 1). We have not reduced the set based on sequence similarity since the proteins might have different conformations based on their ligands. Note, this list might include non-Ebola proteins which might have been co-crystallized with the Ebola protein. However, they have been put through the same analysis since they might provide insights into the Ebola proteins themselves.
Ebola virus envelope protein Minor nucleoprotein VP30 Polymerase cofactor VP35 Membrane-associated protein VP24 Nucleoprotein Matrix protein VP40
These proteins were then analyzed using DSSP15, and resulted in 758 helices in all (See ALPHA.zip in Dataset 1). These helices were then analyzed using PAGAL. The PAGAL algorithm has been detailed previously14. Briefly, the Edmundson wheel is computed by considering a wheel with centre (0,0), radius 5, first residue coordinate (0,5) and advancing each subsequent residue by 100 degrees on the circle, as 3.6 turns of the helix makes one full circle. We compute the hydrophobic moment by connecting the center to the coordinate of the residue and give it a magnitude obtained from the hydrophobic scale (in our case, this scale is obtained from13). These vectors are then added to obtain the final hydrophobic moment.
The color coding is as follows: all hydrophobic residues are colored red, while hydrophilic residues are colored in blue: dark blue for positively charged residues, medium blue for negatively charged residues and light blue for amides.
The raw file generated by analyzing all 146 proteins through PAGAL is provided as PAGALRAWDATA.txt (Dataset 1), and contains the hydrophobic moment, percent of positive charges and the total number of charged residues for every helix. These are then sorted based on the charge (negative or positive) or the hydrophobic moment. We ignore the helices that have none or a single charged residue, and those that are smaller than 10 residues in length. The proportion of charged residues is computed based on the total number of charged residues, and not the length of the helix.
All protein structures were rendered by PyMol (http://www.pymol.org/). The sequence alignment was done using ClustalW23. The alignment images were generated using Seaview24. Protein structures have been superimposed using MUSTANG25.
Results and discussion
Dataset 1.PAGAL analysis of Ebola-related alpha helices.
A PDB database search using the keyword ‘Ebola’ generate 146 single chained proteins, which were analyzed using Define Secondary Structure of Proteins, resulting in 758 alpha helices (ALPHA.zip). Note, this list might include non-Ebola proteins which might have been co-crystallized with the Ebola protein. These helices were analyzed using PAGAL (PAGALRAWDATA.txt), which details the hydrophobic moment, percent of positive charges and the total number of charged residues for every helix.
Helices with large hydrophobic moment
We began by analyzing the helices which have a large hydrophobic moment (hydrophobic scale is obtained from13) (Table 2). The Edmundson wheel for the helix 1EBOE.HELIX1 from the structure of GP2 from the Ebola virus membrane fusion glycoprotein (PDBid:1EBO)16 is shown in Figure 1a. Figure 1b shows the residues comprising these helices (in magenta) in the apo form (PDBid:1EBO)16. The neutralizing antibody (KZ52) derived from a human survivor of the 1995 Kikwit outbreak (PDBid:3CSY)17 recognizes an epitope on this AH, emphasizing the critical nature of this AH in the virulence of the Ebola virus (Figure 1c,d). The antibody most likely inhibits the rearrangement of GP2 segments, which abrogates the fusion of the internal loop in the host membrane17. Table 3 shows the residues in the specified helix (residues 553–597, chain J, PDBid:3CSY) making possible hydrogen bonds with different residues in the human Fab KZ52 heavy chain (residues 1–228, chain A, PDBid:3CSY). Among all the interactions, only Gly553 is on 1EBOE.HELIX1 (at a distance of 2.7 Å from Thr100/OG1), although the others are sequentially proximal. These few interactions are sufficient to bind to this helix, rendering the virus non-virulent, and leading to human recovery. The importance of interfacial hydrophobicity in viral proteins involved in host entry through membrane fusion has recently been discussed in details, and remains ‘an underutilized therapeutic target’26. 1EBOE.HELIX0 (Table 2) also has a high hydrophobic moment, but is actually an isoleucine zipper derived from GCN427 (Figure 1b).
Table 2. Identifying helices with unique properties.
Property based on which the sorting is done is either the Hydrophobic moment (HM) and the percentage of negative (NEG) or positive residues (POS). HM: Hydrophobic moment, RPNR: Ratio of the positive to the negative residues, Len: length of the helix, NCH: number of charged residues. GP: glycoprotein from Ebola, VP24: Membrane-associated protein from Ebola, VP35: Polymerase cofactor.
Property
Protein
Helix
Len
HM
RPNR
NCH
HM
GP GP
1EBOE.HELIX1 1EBOE.HELIX0
46 29
16.2 11.5
0.5 0.5
11 13
NEG
VP24 VP35
4U2XA.HELIX5 3FKEA.HELIX2
16 14
4.4 3.2
0 0.2
2 4
POS
VP24 VP35
4U2XA.HELIX7 3FKEA.HELIX1
19 15
6.5 7.8
0.8 0.8
5 4
Figure 1. Helix with large hydrophobic moment in GP2 from the Ebola virus membrane fusion glycoprotein.
(a) Edmundson wheel for 1EBOE.HELIX1. The hydrophobic moment vector is not to scale. The color coding is as follows: all hydrophobic residues are colored red, while hydrophilic residues are colored in blue: dark blue for positively charged residues, medium blue for negatively charged residues and light blue for amides. (b) Structure of PDBid:1EBOE, 1EBOE.HELIX1 is marked in magenta and the leucine zipper is in blue. (c) 1EBOE.HELIX1 is disrupted by an antibody derived from a human survivor of the 1995 Kikwit outbreak (PDBid:3CSY). (d) Gly553/N on 1EBOE.HELIX1 makes a possible hydrogen bond to Thr100/OG1 at a distance of 2.7 Å.
Table 3. Interactions obtained from the crystal structure of the Ebola virus glycoprotein in complex with a neutralizing antibody from a human survivor.
The helix with a large hydrophobic moment, as determined from PDBid:1EBOE, is disrupted in the structure from PDBid:3CSY through possible hydrogen bonds with different residues in the human Fab KZ52 heavy chain (antibody, chain A). The helix residues are: 553–597 in chain J, PDBid:3CSY.
Helices with high proportion of negatively charged residues. Identifying difference among different species
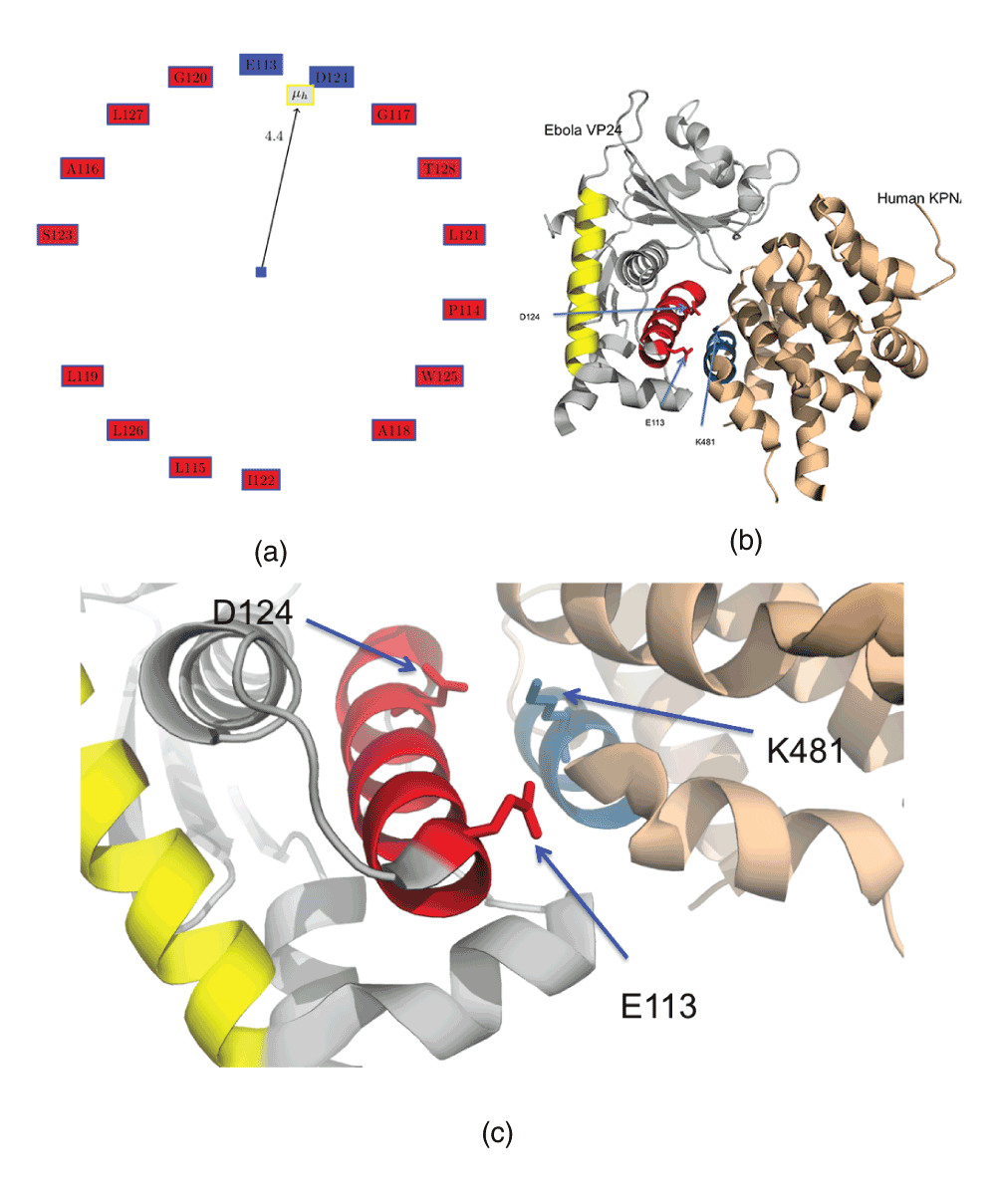
We then analyzed the helices having a high proportion of negatively charged residues, sorted based on the length of the helix when the percentage of negatively residues are the same (Table 2). Figure 2a shows the Edmundson wheel for the helix 4U2XA.HELIX5 (which has only two charged residues - the basic E113 and D124), while Figure 2b,c shows this helix in the protein complex marked in magenta. Protein PDBid:4U2XD is the human karyopherin alpha nuclear transporter (KPNA) C terminus in complex with the Ebola virus VP24 protein (eVP24)18. eVP24 interferes with the immune response by selectively targeting tyrosine-phosphorylated STAT1 nuclear import19. It does not hinder the transport of other cargo that may be required for viral replication. 4U2XA.HELIX5 is responsible for forming the complex with the KPNA protein through a helix (4U2XD.HELIX9, in blue), and K481 from KPNA is in contact with D124 from eVP24 (distance between K481/NZ and D124/OD2 is 3.98 Å). Their interaction is probably electrostatic, since the atoms have opposite charges. VP24 has also been shown to directly bind to STAT1, further compromising the immune response28. Recently, KPNA was docked to Reston Ebola VP24 (PDBid:4D9OA)28 using the VP24 from Zaire Ebola (PDBid:4U2XA)18 as a template29. The docked structure showed that a single mutation might be one of the critical factors responsible for the non-pathogenic nature of Reston Ebola in humans30,31. Also, it was shown that the VP24 from Marburg virus (PDBid:3VNEA)28, which has a different immunosuppressive mechanism than the Ebola virus32, has different properties in the helices responsible for binding KPNA in the Zaire Ebola VP24.
Figure 2. Helix 4U2XA.HELIX5 from membrane-associated protein VP24 with a high proportion of acidic residues.
(a) Edmundson wheel for 4U2XA.HELIX5. (b) Complex of VP24 (PDBid:4U2XA) and human karyopherin alpha nuclear transporters (KPNA) C terminus (PDBid:4U2XD). (c) D124 from VP24 probably has an electrostatic interaction with K481 from KPNA. This interaction is sufficient to interfere with the immune response to Ebola infection.
The next helix having a high proportion of negatively charged residues (3FKEA.HELIX2) is from a VP35, a classic example of a moonlighting protein, that can be a component of the viral RNA polymerase complex, a viral assembly factor, or inhibitor of host interferon production33. We have not been able to identify a critical role for this helix in the protein from current literature. However, VP35 consists of several helices, and is reasonably conserved in the Marburg virus from the same Filoviridae family (42% identity, 58% similarity) (Figure 3a). Often, it is difficult to identify the regions of the protein that differ from a sequence or structural alignment (Figure 3b), in case there is interest in understanding different responses of the proteins to known drugs or even the immune system. Table 4 compares the characteristics of the helices in the VP35 from Ebola and Marburg (the helix numbering is offset by one, due to a small N-terminal helix in the Marburg protein (which might be due to crystallization technique differences and probably is not critical). Thus, we have numbered these helices using alphabets. It can be seen that most of the helices have the same properties, barring helices E and F, where the acidic residue is present in the E helix in Marburg and in the F helix in Ebola. These helices are marked in yellow in Figure 3b. Also, it can be seen that helix C, which has a high proportion of acidic residues in VP35, has a fewer number of those residues in Marburg. The difference in the pathogenicity of these viruses are encoded in the structure of the expressed proteins, and the design of drugs and vaccines to counter virulence should take these differences into account.
Figure 3. Polymerase cofactor VP35 (PDBid:3FKE).
VP35 has several moonlighting functions related to immune evasion. (a) Sequence alignment of VP35 from Marburg (PDBid:4GHLA) and Ebola (PDBid:3FKEA). (b) Structural alignment using MUSTANG. The helices that have differing properties are marked in yellow. 3FKEA.HELIX1 spanning residues 238–252 is marked in magenta. This is a helix with a high proportion of positively charged residues that have been observed to have important interactions in the structure33. (c) Edmundson wheel for 3FKEA.HELIX1.
Table 4. Detecting differences in related proteins based on characteristics of alpha helices.
Comparing the VP35 protein from Marburg (PDBid:4GHLA) and Ebola (PDBid:3FKEA). Note the helices are offset by one, due the presence of an extra helix in the Marburg VP35. Thus, we name the helices using alphabets. It can be seen that most helices have the same properties, barring helices E and F, where the acidic residue is present in the E helix in Marburg and in the F helix in Ebola. HM: Hydrophobic moment, RPNR: Ratio of the positive to the negative residues, Len: length of the helix, NCH: number of charged residues.
Helix Name
Real Helix
Len
HM
RPNR
NCH
A
4GHLA.HELIX1 3FKEA.HELIX0
10 9
2.8 5
0.5 0.7
2 3
B
4GHLA.HELIX2 3FKEA.HELIX1
15 15
5.3 7.8
1 0.8
3 4
C
4GHLA.HELIX3 3FKEA.HELIX2
13 14
4.6 3.2
0.5 0.2
2 4
D
4GHLA.HELIX4 3FKEA.HELIX3
11 11
5.1 3.6
1 1
2 2
E
4GHLA.HELIX5 3FKEA.HELIX4
3 3
2.7 1.1
0 -1
1 0
F
4GHLA.HELIX6 3FKEA.HELIX5
3 3
1 1.3
-1 0.5
0 2
G
4GHLA.HELIX7 3FKEA.HELIX6
6 6
4.7 4.7
1 1
2 2
H
4GHLA.HELIX8 3FKEA.HELIX7
3 3
2.8 3.4
0.5 0.5
2 2
Helices with high proportion of positively charged residues
For helices having a high proportion of positively charged residues, we could not find any reference to the critical nature of the first helix (Table 2, 4U2XA.HELIX7). This helix is marked in yellow in Figure 2c. The second helix (3FKEA.HELIX1) is from VP35, which was discussed previously33. This helix spans residues 238–252 and includes Lys248 and Lys251, a basic patch which is ‘100% identical among members of the Ebola viral isolates’33, and Ala238, Gln241, Leu242, Val245, Ile246, Leu249 which interacts with a β sheet to create a hydrophobic subdomain33. This helix is marked in magenta in Figure 3b, and the Edmundson wheel is shown in Figure 3c. Once again, we demonstrate that unique values of an AH is a strong indicator of its significance in the viral functionality.
Multifunctional/moonlighting
The multifunctional roles played by many of these Ebola proteins is probably due to stretches of intrinsically disordered regions within the structure - ‘fuzzy objects with fuzzy structures and fuzzy functions’34. The conformational plasticity9 and moonlighting abilities of these proteins are key determinants for immune evasion35.
The above examples have analyzed all helices from the Ebola proteome. However, it also possible to analyze the helices in a single protein, and probe those for unique features. Table 5 shows the values obtained from PAGAL for helices of the C-terminal domain of the Zaire Ebola virus nucleoprotein36. It can be seen that 4QAZA.HELIX0 (residues 646–658) has a reasonably high hydrophobic moment (although it will not rank highly if we analyze all helices from the proteome), and also a high number of charged residues (Figure 4a,b). It has been observed that ‘the side chains of Glu645, His646, Glu649, Lys684, Glu695, Glu709, Lys728 and Gln739 are partly disordered so that some or all of their atoms are not visible in the electron density’36. Glu645, His646, Glu649 are part of this helix, and are thus critical to the disorderedness of the protein, which is critical for its moonlighting roles. Note, that Glu has been observed to be the second most disorder promoting residue (after proline)37. Furthermore, Tyr652 and Leu656, which lie in this helix, are residues that have been hypothesized to be part of the protein-protein interaction site involving this protein36.
Table 5. Properties of the helices of the C-terminal domain of the Zaire Ebola virus nucleoprotein (PDBid:4QAZA).
4QAZA.HELIX0 comprising of residues 646–658 has a reasonably large hydrophobic moment, and has been hypothesized to be part of the protein which is involved in protein-protein interactions36. Further, these helices have residues with disordered sidechains36, which are known to be critical for moonlighting functions34. HM: Hydrophobic moment, RPNR: Ratio of the positive to the negative residues, Len: length of the helix, NCH: number of charged residues.
Figure 4. C-terminal domain of the Zaire Ebola virus nucleoprotein36.
(a) Edmundson wheel for 4QAZA.HELIX0 (residues 646–658). (b) Protein structure for PDBid:4QAZA.
Conclusions
The ability of a genome as small as the Ebola virus to inflict a dishearteningly high percentage of mortality in human subjects is a humbling experience in the context of the tremendous technological advancements achieved in the last few decades3,4. The Ebola virus potently suppresses the human immune response2,6,38 by binding with key human proteins involved in the immune pathway18. These protein-protein interactions are often mediated through well structured secondary regions within the protein structures (alpha helices), and the design of molecules that inhibit these ‘hotspots’20,39 has been a well known strategy to develop drugs to counter bacterial and viral infections10–12. For example, synthetic peptides derived from the oligomerization domain of polymerase subunits has been shown to inhibit viral proteins40,41. In addition, there might exist other protein domains that might be exploited by non-native viral peptides to obstruct viral functionality. In the current work, we characterize alpha helices in the Ebola virus proteome using a recently implemented open access software (PAGAL)14, thus identifying potential targets for inhibition of the helix mediated interactions. Through several examples, we demonstrate that helices with unique features are involved in interactions with host proteins (either antibodies from survivors, or proteins regulating the immune response). Further, we also provide an alternate way of analyzing differences in related proteins (from the Marburg virus) by focusing on the properties of corresponding helices. As future work, we intend to develop methodologies to design peptides that would target these ‘hotspots’39. It has to be kept in mind that it has been a challenge to design small ligands that disrupt protein-protein interactions, and designers resort to several innovative techniques to overcome thermodynamic instability or proteolytic susceptibility42–45. These helices can essentially be epitopes46,47 for developing antibodies against the virus48,49. Interestingly, ZMapp, a cocktail of three antibodies has shown reversion of advanced Ebola symptoms in non-human primates50, and uses only glycoprotein-specific epitope generated antibodies47,51. It is interesting to hypothesize that additions to this cocktail with antibodies derived from other epitopes (for example, 4U2XA.HELIX5 from VP24 that is involved in immune response suppression) could prove more effective. Thus, we provide a comprehensive list of potential targets within the small proteome of the Ebola virus that can directed rational design to quickly innovate therapies.
SC wrote the computer programs. All authors analyzed the data, and contributed equally to the writing and subsequent refinement of the manuscript.
Competing interests
No competing interests were disclosed.
Grant information
AMD wishes to acknowledge grant support from the California Department of Food and Agriculture PD/GWSS Board. BJ acknowledges financial support from Tata Institute of Fundamental Research (Department of Atomic Energy). Additionally, BJR is thankful to the Department of Science and Technology for the JC Bose Award Grant. BA acknowledges financial support from the Science Institute of the University of Iceland.
Faculty Opinions recommended
References
1.
Pattyn S, van der Groen G, Courteille G, et al.:
Isolation of Marburg-like virus from a case of haemorrhagic fever in Zaire.
Lancet.
1977; 1(8011): 573–574. PubMed Abstract
| Publisher Full Text
4.
Piot P, Muyembe JJ, Edmunds WJ:
Ebola in west Africa: from disease outbreak to humanitarian crisis.
Lancet Infect Dis.
2014; 14(11): 1034–1035. PubMed Abstract
| Publisher Full Text
5.
Kiley M, Bowen E, Eddy G, et al.:
Filoviridae: a taxonomic home for Marburg and Ebola Viruses?
Intervirology.
1982; 18(1–2): 24–32. PubMed Abstract
| Publisher Full Text
6.
Daugherty MD, Malik HS:
How a virus blocks a cellular emergency access lane to the nucleus, STAT!
Cell Host Microbe.
2014; 16(2): 150–152. PubMed Abstract
| Publisher Full Text
7.
Elliott LH, Kiley MP, McCormick JB:
Descriptive analysis of Ebola virus proteins.
Virology.
1985; 147(1): 169–176. PubMed Abstract
| Publisher Full Text
8.
Bornholdt ZA, Noda T, Abelson DM, et al.:
Structural basis for ebolavirus matrix assembly and budding; protein plasticity allows multiple functions.
Cell.
2013; 154: 763. PubMed Abstract
| Publisher Full Text
| Free Full Text
9.
Radzimanowski J, Effantin G, Weissenhorn W:
Conformational plasticity of the Ebola virus matrix protein.
Protein Sci.
2014; 23(11): 1519–1527. PubMed Abstract
| Publisher Full Text
10.
Wild CT, Shugars DC, Greenwell TK, et al.:
Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection.
Proc Natl Acad Sci U S A.
1994; 91(21): 9770–9774. PubMed Abstract
| Publisher Full Text
| Free Full Text
11.
Judice JK, Tom JY, Huang W, et al.:
Inhibition of HIV type 1 infectivity by constrained alpha-helical peptides: implications for the viral fusion mechanism.
Proc Natl Acad Sci U S A.
1997; 94(25): 13426–13430. PubMed Abstract
| Publisher Full Text
| Free Full Text
12.
Ernst JT, Kutzki O, Debnath AK, et al.:
Design of a protein surface antagonist based on α-helix mimicry: Inhibition of gp41 assembly and viral fusion.
Angew Chem Int Ed Engl.
2002; 41(2): 278–281. PubMed Abstract
| Publisher Full Text
13.
Jones MK, Anantharamaiah GM, Segrest JP:
Computer programs to identify and classify amphipathic alpha helical domains.
J Lipid Res.
1992; 33(2): 287–296. PubMed Abstract
15.
Joosten RP, te Beek TA, Krieger E, et al.:
A series of PDB related databases for everyday needs.
Nucleic Acids Res.
2011; 39(Database issue): D411–419. PubMed Abstract
| Publisher Full Text
| Free Full Text
16.
Weissenhorn W, Carfi A, Lee KH, et al.:
Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain.
Mol Cell.
1998; 2(5): 605–616. PubMed Abstract
| Publisher Full Text
17.
Lee JE, Fusco ML, Hessell AJ, et al.:
Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor.
Nature.
2008; 454(7201): 177–182. PubMed Abstract
| Publisher Full Text
| Free Full Text
18.
Xu W, Edwards MR, Borek DM, et al.:
Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1.
Cell Host Microbe.
2014; 16(2): 187–200. PubMed Abstract
| Publisher Full Text
| Free Full Text
19.
Reid SP, Leung LW, Hartman AL, et al.:
Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation.
J Virol.
2006; 80(11): 5156–5167. PubMed Abstract
| Publisher Full Text
| Free Full Text
20.
Azzarito V, Long K, Murphy NS, et al.:
Inhibition of α-helix-mediated protein-protein interactions using designed molecules.
Nat Chem.
2013; 5(3): 161–173. PubMed Abstract
| Publisher Full Text
21.
Suzuki Y, Gojobori T:
The origin and evolution of ebola and marburg viruses.
Mol Biol Evol.
1997; 14(8): 800–806. PubMed Abstract
| Publisher Full Text
22.
Feldmann H, Nichol ST, Klenk HD, et al.:
Characterization of filoviruses based on differences in structure and antigenicity of the virion glycoprotein.
Virology.
1994; 199(2): 469–473. PubMed Abstract
| Publisher Full Text
23.
Larkin MA, Blackshields G, Brown NP, et al.:
Clustal W and Clustal X version 2.0.
Bioinformatics.
2007; 23(21): 2947–2948. PubMed Abstract
| Publisher Full Text
24.
Gouy M, Guindon S, Gascuel O:
SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building.
Mol Biol Evol.
2010; 27(2): 221–224. PubMed Abstract
| Publisher Full Text
25.
Konagurthu AS, Whisstock JC, Stuckey PJ, et al.:
MUSTANG: a multiple structural alignment algorithm.
Proteins.
2006; 64(3): 559–574. PubMed Abstract
| Publisher Full Text
26.
Badani H, Garry RF, Wimley WC:
Peptide entry inhibitors of enveloped viruses: the importance of interfacial hydrophobicity.
Biochim Biophys Acta.
2014; 1838(9): 2180–97. PubMed Abstract
| Publisher Full Text
27.
Weissenhorn W, Calder LJ, Wharton SA, et al.:
The central structural feature of the membrane fusion protein subunit from the Ebola virus glycoprotein is a long triple-stranded coiled coil.
Proc Natl Acad Sci U S A.
1998; 95(11): 6032–6036. PubMed Abstract
| Publisher Full Text
| Free Full Text
28.
Zhang AP, Bornholdt ZA, Liu T, et al.:
The ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold.
PLoS Pathog.
2012; 8(2): e1002550. PubMed Abstract
| Publisher Full Text
| Free Full Text
29.
Chakraborty S, Rao B, Asgeirsson B, et al.:
Correlating the ability of VP24 protein from Ebola and Marburg viruses to bind human karyopherin to their immune suppression mechanism and pathogenicity using computational methods [v1; ref status: awaiting peer review, http://f1000r.es/4o3].
F1000Res.
2014; 3: 265. Publisher Full Text
30.
Miranda ME, White ME, Dayrit MM, et al.:
Seroepidemiological study of filovirus related to Ebola in the Philippines.
Lancet.
1991; 337(8738): 425–426. PubMed Abstract
| Publisher Full Text
31.
Miranda ME, Miranda NL:
Reston ebolavirus in humans and animals in the Philippines: a review.
J Infect Dis.
2011; 204(Suppl 3): S757–S760. PubMed Abstract
| Publisher Full Text
32.
Valmas C, Grosch MN, Schumann M, et al.:
Marburg virus evades interferon responses by a mechanism distinct from Ebola virus.
PLoS Pathog.
2010; 6(1): e1000721. PubMed Abstract
| Publisher Full Text
| Free Full Text
33.
Leung DW, Ginder ND, Fulton DB, et al.:
Structure of the Ebola VP35 interferon inhibitory domain.
Proc Natl Acad Sci U S A.
2009; 106(2): 411–416. PubMed Abstract
| Publisher Full Text
| Free Full Text
34.
Uversky VN:
Intrinsically disordered proteins from A to Z.
Int J Biochem Cell Biol.
2011; 43(8): 1090–1103. PubMed Abstract
| Publisher Full Text
36.
Dziubanska PJ, Derewenda U, Ellena JF, et al.:
The structure of the C-terminal domain of the Zaire ebolavirus nucleoprotein.
Acta Crystallogr D Biol Crystallogr.
2014; 70(Pt 9): 2420–2429. PubMed Abstract
| Publisher Full Text
| Free Full Text
37.
Uversky VN:
The alphabet of intrinsic disorder: II. various roles of glutamic acid in ordered and intrinsically disordered proteins.
Intrinsically Disord Proteins.
2013; 1: 18–40. Publisher Full Text
38.
Kash JC, Mϋhlberger E, Carter V, et al.:
Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type i interferon response is associated with enhanced virulence.
J Virol.
2006; 80(6): 3009–3020. PubMed Abstract
| Publisher Full Text
| Free Full Text
39.
Wells JA, McClendon CL:
Reaching for high-hanging fruit in drug discovery at protein-protein interfaces.
Nature.
2007; 450(7172): 1001–1009. PubMed Abstract
| Publisher Full Text
40.
Hartlieb B, Modrof J, Mϋhlberger E, et al.:
Oligomerization of Ebola virus VP30 is essential for viral transcription and can be inhibited by a synthetic peptide.
J Biol Chem.
2003; 278(43): 41830–41836. PubMed Abstract
| Publisher Full Text
42.
Chapman RN, Dimartino G, Arora PS:
A highly stable short alpha-helix constrained by a main-chain hydrogen-bond surrogate.
J Am Chem Soc.
2004; 126(39): 12252–12253. PubMed Abstract
| Publisher Full Text
43.
Bird GH, Madani N, Perry AF, et al.:
Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic.
Proc Natl Acad Sci U S A.
2010; 107(32): 14093–14098. PubMed Abstract
| Publisher Full Text
| Free Full Text
44.
Bird GH, Boyapalle S, Wong T, et al.:
Mucosal delivery of a double-stapled RSV peptide prevents nasopulmonary infection.
J Clin Invest.
2014; 124(5): 2113–24. PubMed Abstract
| Publisher Full Text
| Free Full Text
45.
Harrison RS, Shepherd NE, Hoang HN, et al.:
Downsizing human, bacterial, and viral proteins to short water-stable alpha helices that maintain biological potency.
Proc Natl Acad Sci U S A.
2010; 107(26): 11686–11691. PubMed Abstract
| Publisher Full Text
| Free Full Text
46.
Takada A, Feldmann H, Stroeher U, et al.:
Identification of protective epitopes on ebola virus glycoprotein at the single amino acid level by using recombinant vesicular stomatitis viruses.
J Virol.
2003; 77(2): 1069–1074. PubMed Abstract
| Publisher Full Text
| Free Full Text
47.
Wilson JA, Hevey M, Bakken R, et al.:
Epitopes involved in antibody-mediated protection from Ebola virus.
Science.
2000; 287(5458): 1664–1666. PubMed Abstract
| Publisher Full Text
48.
Takada A, Ebihara H, Jones S, et al.:
Protective efficacy of neutralizing antibodies against Ebola Virus infection.
Vaccine.
2007; 25(6): 993–999. PubMed Abstract
| Publisher Full Text
49.
Qiu X, Alimonti JB, Melito PL, et al.:
Characterization of Zaire ebolavirus. glycoprotein-specific monoclonal antibodies.
Clin Immunol.
2011; 141(2): 218–227. PubMed Abstract
| Publisher Full Text
51.
Olinger GG Jr, Pettitt J, Kim D, et al.:
Delayed treatment of Ebola virus infection with plant-derived monoclonal antibodies provides protection in rhesus macaques.
Proc Natl Acad Sci U S A.
2012; 109(44): 18030–18035. PubMed Abstract
| Publisher Full Text
| Free Full Text
52.
Chakraborty S, Rao BJ, Asgeirsson B, et al.:
Dataset 1. PAGAL analysis of Ebola-related alpha helices.
F1000Research.
2014. Data Source
1
Plant Sciences Department, University of California, Davis, 95616, USA 2
Department of Biological Sciences, Tata Institute of Fundamental Research, Homi Bhabha Road, Mumbai, 400 005, India 3
Science Institute, Department of Biochemistry, University of Iceland, Dunhaga 3, IS-107 Reykjavik, Iceland
AMD wishes to acknowledge grant support from the California Department of Food and Agriculture PD/GWSS Board. BJ acknowledges financial support from Tata Institute of Fundamental Research (Department of Atomic Energy). Additionally, BJR is thankful to the Department of Science and Technology for the JC Bose Award Grant. BA acknowledges financial support from the Science Institute of the University of Iceland.
Chakraborty S, Rao BJ, Asgeirsson B and Dandekar AM. Characterizing alpha helical properties of Ebola viral proteins as potential targets for inhibition of alpha-helix mediated protein-protein interactions [version 2; peer review: 1 approved]. F1000Research 2014, 3:251 (https://doi.org/10.12688/f1000research.5573.2)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
The authors have responded to my previous questions and improved the manuscript. Although, the described computational approach can identify alpha helices with unique features, the author’s proposal that the helical propensities can be linked to host protein interactions is rather
... Continue reading
The authors have responded to my previous questions and improved the manuscript. Although, the described computational approach can identify alpha helices with unique features, the author’s proposal that the helical propensities can be linked to host protein interactions is rather weak and requires experimental data to validate the method.
There are still a conceptual error in the manuscript:
Page 4: "The antibody most likely inhibits the rearrangement of GP2 segments, which abrogates the fusion of the internal loop in the host membrane."
Do the authors mean that the antibody binding prevents Gp2 conformational changes required for membrane fusion?
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Sandeep Chakraborty, Tata Institute of Fundamental Research, India
27 Jan 2015
Author Response
Dear Dr Weissenhorn, We appreciate your positive comments to the changes we had made in accordance to your previous suggestions. By the statement `The antibody most likely inhibits the rearrangement of
...
Continue readingDear Dr Weissenhorn, We appreciate your positive comments to the changes we had made in accordance to your previous suggestions. By the statement `The antibody most likely inhibits the rearrangement of GP2 segments, which abrogates the fusion of the internal loop in the host membrane.', we meant the same thing as `antibody binding prevents Gp2 conformational changes required for membrane fusion', which is the accepted hypothesis. We could not detect any difference in the meaning of the two wordings.
Please also find a new version of the manuscript which includes further corroboration of our hypothesis. best regards, Sandeep
Dear Dr Weissenhorn, We appreciate your positive comments to the changes we had made in accordance to your previous suggestions. By the statement `The antibody most likely inhibits the rearrangement of GP2 segments, which abrogates the fusion of the internal loop in the host membrane.', we meant the same thing as `antibody binding prevents Gp2 conformational changes required for membrane fusion', which is the accepted hypothesis. We could not detect any difference in the meaning of the two wordings.
Please also find a new version of the manuscript which includes further corroboration of our hypothesis. best regards, Sandeep
Competing Interests:No competing interests were disclosed.Close
Sandeep Chakraborty, Tata Institute of Fundamental Research, India
27 Jan 2015
Author Response
Dear Dr Weissenhorn, We appreciate your positive comments to the changes we had made in accordance to your previous suggestions. By the statement `The antibody most likely inhibits the rearrangement of
...
Continue readingDear Dr Weissenhorn, We appreciate your positive comments to the changes we had made in accordance to your previous suggestions. By the statement `The antibody most likely inhibits the rearrangement of GP2 segments, which abrogates the fusion of the internal loop in the host membrane.', we meant the same thing as `antibody binding prevents Gp2 conformational changes required for membrane fusion', which is the accepted hypothesis. We could not detect any difference in the meaning of the two wordings.
Please also find a new version of the manuscript which includes further corroboration of our hypothesis. best regards, Sandeep
Dear Dr Weissenhorn, We appreciate your positive comments to the changes we had made in accordance to your previous suggestions. By the statement `The antibody most likely inhibits the rearrangement of GP2 segments, which abrogates the fusion of the internal loop in the host membrane.', we meant the same thing as `antibody binding prevents Gp2 conformational changes required for membrane fusion', which is the accepted hypothesis. We could not detect any difference in the meaning of the two wordings.
Please also find a new version of the manuscript which includes further corroboration of our hypothesis. best regards, Sandeep
Competing Interests:No competing interests were disclosed.Close
The authors suggest that they can identify alpha helices and predict their propensities to be targeted by small molecules. Their test case is the small Ebola virus genome, where several crystal structures are available.
First they compute the hydrophobic moment of
... Continue reading
The authors suggest that they can identify alpha helices and predict their propensities to be targeted by small molecules. Their test case is the small Ebola virus genome, where several crystal structures are available.
First they compute the hydrophobic moment of identified helices with their previously published program PAGAL and classify them based on hydrophobicity, positive or negative charges. They conclude that helices with unique feature values are involved in host protein interaction.
Page 4: It is not correct to state that “ this helix is disrupted by a neutralizing antibody derived from a human survivor …”. HR1 or helix 1 from Gp2 is split into 4 small helices in the native GP structure and antibody binding prevents its refolding into the post fusion conformation represented by the Gp2 structure. Now one can argue that small molecules could interfere with the formation of the triple stranded coiled coil formed by HR1 in the post fusion structure. This needs to be clarified in the text.
Next they identified a charged helix in Vps24 that interacts with karyopherin. Why was this chosen? Because of the available structure? This helix contains only two charged residues and would not fall under the classification of carrying a high charge!
The third helices described in detail are from Vps35 and the authors identify several helices with carry charges, but no clear targets are discussed.
Page 6: The authors make a connection between the number of acidic residues in a helix from Ebola Vps35 compared to Marburg Vps35 and the frequency of outbreaks, which is a complete over interpretation of their data.
In summary the manuscript describes an interesting approach to identify or validate potential drug targets. However, the authors need to be more cautious in interpreting their results. Without any experimental validation their approach to link helical properties to protein interaction propensities is extremely weak.
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard, however I have significant reservations, as outlined above.
Sandeep Chakraborty, Tata Institute of Fundamental Research, India
06 Nov 2014
Author Response
Dear Dr Weissenhorn,
We would like to thank you for taking the time to review this paper, and for your suggestions to improve the manuscript. In the interim period, we have
...
Continue readingDear Dr Weissenhorn,
We would like to thank you for taking the time to review this paper, and for your suggestions to improve the manuscript. In the interim period, we have applied other computational methods[ref-1] to correlate the different immunosuppressive and pathogenicity mechanisms in Ebola and Marburg viruses to variations in their structures/sequences [ref-2]. Please find our detailed responses to your comments below.
The authors suggest that they can identify alpha helices and predict their propensities to be targeted by small molecules. Their test case is the small Ebola virus genome, where several crystal structures are available. First they compute the hydrophobic moment of identified helices with their previously published program PAGAL and classify them based on hydrophobicity, positive or negative charges. They conclude that helices with unique feature values are involved in host protein interaction. Page 4: It is not correct to state that this helix is disrupted by a neutralizing anti- body derived from a human survivor . HR1 or helix 1 from Gp2 is split into 4 small helices in the native GP structure and antibody binding prevents its refolding into the post fusion conformation represented by the Gp2 structure. Now one can argue that small molecules could interfere with the formation of the triple stranded coiled coil formed by HR1 in the post fusion structure. This needs to be clarified in the text.
We appreciate this point, (‘KZ52 likely neutralizes by preventing rearrangement of the GP2 HR1A/HR1B segments and blocking host membrane insertion of the internal fusion loop’ [ref-3]), and have made the correction.
Next they identified a charged helix in Vps24 that interacts with karyopherin. Why was this chosen? Because of the available structure? This helix contains only two charged residues and would not fall under the classification of carrying a high charge!
VP24 came up in the sorted list since it has a ‘high proportion of negatively charged residues’, and not high charge. The proportion of charged residues is computed based on the total number of charged residues, and not the length of the helix. We could also create a category of high charge by combining the previous feature (high proportion) to high number of charged residues.
Our search criteria excludes AHs with zero or one charged residue. We had stated this in the Methods section - We ignore the helices that have none or a single charged residue. We also had a cutoff on the length of the AH as 10 - i.e. we are looking for reasonably long AHs - we had not mentioned this constraint. We have modified the Methods section to reflect this. An AH having just two similarly charged residues in a reasonably long AH (and none other) is relatively significant. For example, one charged residue in VP24 (D124) makes an electrostatic contact with human karyopherin, while the other one E113 makes a contact to Arg140 in another helix (α6) in VP24 [ref-2].
The third helices described in detail are from Vps35 and the authors identify several helices with carry charges, but no clear targets are discussed.
We have stated that ‘we have not been able to identify a critical role for this helix in the protein from current literature’, which does not preclude the importance of these helices. This, in fact, highlights the ability of our method to extract helices that might be of significance, yet not probed sufficiently as targets. At the same time, it is also equally possible that this helix is not functionally significant.
Page 6: The authors make a connection between the number of acidic residues in ahelix from Ebola Vps35 compared to Marburg Vps35 and the frequency of outbreaks, which is a complete over interpretation of their data. We agree with this criticism, and have made the corrections.
In summary the manuscript describes an interesting approach to identify or validate potential drug targets.
We appreciate the positive and encouraging note on our efforts to use computational methods to identify critical regions of interaction in the Ebola proteins, which could be easily extended to other organisms as well.
However, the authors need to be more cautious in interpreting their results. Without any experimental validation their approach to link helical properties to protein interaction propensities is extremely weak.
We hope that we have addressed your concerns by the changes that we have made. We also expect future results to corroborate some of our predictions, and will make the updates on the f1000 site (which their format allows us to). We sincerely hope that the manuscript will be found suitable in the modified form for publication.
Thanking you,
Sincerely,
Sandeep Chakraborty (Corresponding author)
[References]
[[1|type=journal|title=DOCLASP - Docking ligands to target proteins using spatial and electrostatic congruence extracted from a known holoenzyme and applying simple geometrical transformations [v1; ref status: awaiting peer review, http://f1000r.es/48g]|authors=Chakraborty/S|source=F1000Research|year=2014|vol=3|issue=262|doi=10.12688/f1000research.5145.1|url=http://f1000research.com/articles/3-262/v1]] [[2|type=journal|title=Correlating the ability of VP24 protein from Ebola and Marburg viruses to bind human karyopherin to their immune suppression mechanism and pathogenicity using computational methods [v1; ref status: awaiting peer review, http://f1000r.es/4o3]|authors=Chakraborty/S|source=F1000Research|year=2014|vol=3|issue=265|doi=10.12688/f1000research.5666.1|url=http://f1000research.com/articles/3-265/v1]] [[3|type=journal|title=Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor|authors=Lee/JE;Fusco/ML;Hessell/AJ;Oswald/EB;Burton/DR;Saphire/EO|source=Nature|year=2008|vol=454|issue=7201|fpage=177|lpage=182|pmid=18615077|pmcid=2700032|doi=10.1038/nature07082|url=http://www.nature.com/nature/journal/v454/n7201/full/nature07082.html]]
Dear Dr Weissenhorn,
We would like to thank you for taking the time to review this paper, and for your suggestions to improve the manuscript. In the interim period, we have applied other computational methods1 to correlate the different immunosuppressive and pathogenicity mechanisms in Ebola and Marburg viruses to variations in their structures/sequences 2. Please find our detailed responses to your comments below.
The authors suggest that they can identify alpha helices and predict their propensities to be targeted by small molecules. Their test case is the small Ebola virus genome, where several crystal structures are available. First they compute the hydrophobic moment of identified helices with their previously published program PAGAL and classify them based on hydrophobicity, positive or negative charges. They conclude that helices with unique feature values are involved in host protein interaction. Page 4: It is not correct to state that this helix is disrupted by a neutralizing anti- body derived from a human survivor . HR1 or helix 1 from Gp2 is split into 4 small helices in the native GP structure and antibody binding prevents its refolding into the post fusion conformation represented by the Gp2 structure. Now one can argue that small molecules could interfere with the formation of the triple stranded coiled coil formed by HR1 in the post fusion structure. This needs to be clarified in the text.
We appreciate this point, (‘KZ52 likely neutralizes by preventing rearrangement of the GP2 HR1A/HR1B segments and blocking host membrane insertion of the internal fusion loop’ 3), and have made the correction.
Next they identified a charged helix in Vps24 that interacts with karyopherin. Why was this chosen? Because of the available structure? This helix contains only two charged residues and would not fall under the classification of carrying a high charge!
VP24 came up in the sorted list since it has a ‘high proportion of negatively charged residues’, and not high charge. The proportion of charged residues is computed based on the total number of charged residues, and not the length of the helix. We could also create a category of high charge by combining the previous feature (high proportion) to high number of charged residues.
Our search criteria excludes AHs with zero or one charged residue. We had stated this in the Methods section - We ignore the helices that have none or a single charged residue. We also had a cutoff on the length of the AH as 10 - i.e. we are looking for reasonably long AHs - we had not mentioned this constraint. We have modified the Methods section to reflect this. An AH having just two similarly charged residues in a reasonably long AH (and none other) is relatively significant. For example, one charged residue in VP24 (D124) makes an electrostatic contact with human karyopherin, while the other one E113 makes a contact to Arg140 in another helix (α6) in VP24 2.
The third helices described in detail are from Vps35 and the authors identify several helices with carry charges, but no clear targets are discussed.
We have stated that ‘we have not been able to identify a critical role for this helix in the protein from current literature’, which does not preclude the importance of these helices. This, in fact, highlights the ability of our method to extract helices that might be of significance, yet not probed sufficiently as targets. At the same time, it is also equally possible that this helix is not functionally significant.
Page 6: The authors make a connection between the number of acidic residues in ahelix from Ebola Vps35 compared to Marburg Vps35 and the frequency of outbreaks, which is a complete over interpretation of their data. We agree with this criticism, and have made the corrections.
In summary the manuscript describes an interesting approach to identify or validate potential drug targets.
We appreciate the positive and encouraging note on our efforts to use computational methods to identify critical regions of interaction in the Ebola proteins, which could be easily extended to other organisms as well.
However, the authors need to be more cautious in interpreting their results. Without any experimental validation their approach to link helical properties to protein interaction propensities is extremely weak.
We hope that we have addressed your concerns by the changes that we have made. We also expect future results to corroborate some of our predictions, and will make the updates on the f1000 site (which their format allows us to). We sincerely hope that the manuscript will be found suitable in the modified form for publication.
Thanking you,
Sincerely,
Sandeep Chakraborty (Corresponding author)
References 1. Chakraborty S: DOCLASP - Docking ligands to target proteins using spatial and electrostatic congruence extracted from a known holoenzyme and applying simple geometrical transformations [v1; ref status: awaiting peer review, http://f1000r.es/48g]. F1000Research. 2014; 3 (262). Publisher Full Text | Reference Source 2. Chakraborty S: Correlating the ability of VP24 protein from Ebola and Marburg viruses to bind human karyopherin to their immune suppression mechanism and pathogenicity using computational methods [v1; ref status: awaiting peer review, http://f1000r.es/4o3]. F1000Research. 2014; 3 (265). Publisher Full Text | Reference Source 3. Lee JE, Fusco ML, Hessell AJ, Oswald EB, et al.: Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008; 454 (7201): 177-182 PubMed Abstract | Free Full Text | Publisher Full Text | Reference Source
Competing Interests:No competing interests were disclosed.Close
Sandeep Chakraborty, Tata Institute of Fundamental Research, India
06 Nov 2014
Author Response
Dear Dr Weissenhorn,
We would like to thank you for taking the time to review this paper, and for your suggestions to improve the manuscript. In the interim period, we have
...
Continue readingDear Dr Weissenhorn,
We would like to thank you for taking the time to review this paper, and for your suggestions to improve the manuscript. In the interim period, we have applied other computational methods[ref-1] to correlate the different immunosuppressive and pathogenicity mechanisms in Ebola and Marburg viruses to variations in their structures/sequences [ref-2]. Please find our detailed responses to your comments below.
The authors suggest that they can identify alpha helices and predict their propensities to be targeted by small molecules. Their test case is the small Ebola virus genome, where several crystal structures are available. First they compute the hydrophobic moment of identified helices with their previously published program PAGAL and classify them based on hydrophobicity, positive or negative charges. They conclude that helices with unique feature values are involved in host protein interaction. Page 4: It is not correct to state that this helix is disrupted by a neutralizing anti- body derived from a human survivor . HR1 or helix 1 from Gp2 is split into 4 small helices in the native GP structure and antibody binding prevents its refolding into the post fusion conformation represented by the Gp2 structure. Now one can argue that small molecules could interfere with the formation of the triple stranded coiled coil formed by HR1 in the post fusion structure. This needs to be clarified in the text.
We appreciate this point, (‘KZ52 likely neutralizes by preventing rearrangement of the GP2 HR1A/HR1B segments and blocking host membrane insertion of the internal fusion loop’ [ref-3]), and have made the correction.
Next they identified a charged helix in Vps24 that interacts with karyopherin. Why was this chosen? Because of the available structure? This helix contains only two charged residues and would not fall under the classification of carrying a high charge!
VP24 came up in the sorted list since it has a ‘high proportion of negatively charged residues’, and not high charge. The proportion of charged residues is computed based on the total number of charged residues, and not the length of the helix. We could also create a category of high charge by combining the previous feature (high proportion) to high number of charged residues.
Our search criteria excludes AHs with zero or one charged residue. We had stated this in the Methods section - We ignore the helices that have none or a single charged residue. We also had a cutoff on the length of the AH as 10 - i.e. we are looking for reasonably long AHs - we had not mentioned this constraint. We have modified the Methods section to reflect this. An AH having just two similarly charged residues in a reasonably long AH (and none other) is relatively significant. For example, one charged residue in VP24 (D124) makes an electrostatic contact with human karyopherin, while the other one E113 makes a contact to Arg140 in another helix (α6) in VP24 [ref-2].
The third helices described in detail are from Vps35 and the authors identify several helices with carry charges, but no clear targets are discussed.
We have stated that ‘we have not been able to identify a critical role for this helix in the protein from current literature’, which does not preclude the importance of these helices. This, in fact, highlights the ability of our method to extract helices that might be of significance, yet not probed sufficiently as targets. At the same time, it is also equally possible that this helix is not functionally significant.
Page 6: The authors make a connection between the number of acidic residues in ahelix from Ebola Vps35 compared to Marburg Vps35 and the frequency of outbreaks, which is a complete over interpretation of their data. We agree with this criticism, and have made the corrections.
In summary the manuscript describes an interesting approach to identify or validate potential drug targets.
We appreciate the positive and encouraging note on our efforts to use computational methods to identify critical regions of interaction in the Ebola proteins, which could be easily extended to other organisms as well.
However, the authors need to be more cautious in interpreting their results. Without any experimental validation their approach to link helical properties to protein interaction propensities is extremely weak.
We hope that we have addressed your concerns by the changes that we have made. We also expect future results to corroborate some of our predictions, and will make the updates on the f1000 site (which their format allows us to). We sincerely hope that the manuscript will be found suitable in the modified form for publication.
Thanking you,
Sincerely,
Sandeep Chakraborty (Corresponding author)
[References]
[[1|type=journal|title=DOCLASP - Docking ligands to target proteins using spatial and electrostatic congruence extracted from a known holoenzyme and applying simple geometrical transformations [v1; ref status: awaiting peer review, http://f1000r.es/48g]|authors=Chakraborty/S|source=F1000Research|year=2014|vol=3|issue=262|doi=10.12688/f1000research.5145.1|url=http://f1000research.com/articles/3-262/v1]] [[2|type=journal|title=Correlating the ability of VP24 protein from Ebola and Marburg viruses to bind human karyopherin to their immune suppression mechanism and pathogenicity using computational methods [v1; ref status: awaiting peer review, http://f1000r.es/4o3]|authors=Chakraborty/S|source=F1000Research|year=2014|vol=3|issue=265|doi=10.12688/f1000research.5666.1|url=http://f1000research.com/articles/3-265/v1]] [[3|type=journal|title=Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor|authors=Lee/JE;Fusco/ML;Hessell/AJ;Oswald/EB;Burton/DR;Saphire/EO|source=Nature|year=2008|vol=454|issue=7201|fpage=177|lpage=182|pmid=18615077|pmcid=2700032|doi=10.1038/nature07082|url=http://www.nature.com/nature/journal/v454/n7201/full/nature07082.html]]
Dear Dr Weissenhorn,
We would like to thank you for taking the time to review this paper, and for your suggestions to improve the manuscript. In the interim period, we have applied other computational methods1 to correlate the different immunosuppressive and pathogenicity mechanisms in Ebola and Marburg viruses to variations in their structures/sequences 2. Please find our detailed responses to your comments below.
The authors suggest that they can identify alpha helices and predict their propensities to be targeted by small molecules. Their test case is the small Ebola virus genome, where several crystal structures are available. First they compute the hydrophobic moment of identified helices with their previously published program PAGAL and classify them based on hydrophobicity, positive or negative charges. They conclude that helices with unique feature values are involved in host protein interaction. Page 4: It is not correct to state that this helix is disrupted by a neutralizing anti- body derived from a human survivor . HR1 or helix 1 from Gp2 is split into 4 small helices in the native GP structure and antibody binding prevents its refolding into the post fusion conformation represented by the Gp2 structure. Now one can argue that small molecules could interfere with the formation of the triple stranded coiled coil formed by HR1 in the post fusion structure. This needs to be clarified in the text.
We appreciate this point, (‘KZ52 likely neutralizes by preventing rearrangement of the GP2 HR1A/HR1B segments and blocking host membrane insertion of the internal fusion loop’ 3), and have made the correction.
Next they identified a charged helix in Vps24 that interacts with karyopherin. Why was this chosen? Because of the available structure? This helix contains only two charged residues and would not fall under the classification of carrying a high charge!
VP24 came up in the sorted list since it has a ‘high proportion of negatively charged residues’, and not high charge. The proportion of charged residues is computed based on the total number of charged residues, and not the length of the helix. We could also create a category of high charge by combining the previous feature (high proportion) to high number of charged residues.
Our search criteria excludes AHs with zero or one charged residue. We had stated this in the Methods section - We ignore the helices that have none or a single charged residue. We also had a cutoff on the length of the AH as 10 - i.e. we are looking for reasonably long AHs - we had not mentioned this constraint. We have modified the Methods section to reflect this. An AH having just two similarly charged residues in a reasonably long AH (and none other) is relatively significant. For example, one charged residue in VP24 (D124) makes an electrostatic contact with human karyopherin, while the other one E113 makes a contact to Arg140 in another helix (α6) in VP24 2.
The third helices described in detail are from Vps35 and the authors identify several helices with carry charges, but no clear targets are discussed.
We have stated that ‘we have not been able to identify a critical role for this helix in the protein from current literature’, which does not preclude the importance of these helices. This, in fact, highlights the ability of our method to extract helices that might be of significance, yet not probed sufficiently as targets. At the same time, it is also equally possible that this helix is not functionally significant.
Page 6: The authors make a connection between the number of acidic residues in ahelix from Ebola Vps35 compared to Marburg Vps35 and the frequency of outbreaks, which is a complete over interpretation of their data. We agree with this criticism, and have made the corrections.
In summary the manuscript describes an interesting approach to identify or validate potential drug targets.
We appreciate the positive and encouraging note on our efforts to use computational methods to identify critical regions of interaction in the Ebola proteins, which could be easily extended to other organisms as well.
However, the authors need to be more cautious in interpreting their results. Without any experimental validation their approach to link helical properties to protein interaction propensities is extremely weak.
We hope that we have addressed your concerns by the changes that we have made. We also expect future results to corroborate some of our predictions, and will make the updates on the f1000 site (which their format allows us to). We sincerely hope that the manuscript will be found suitable in the modified form for publication.
Thanking you,
Sincerely,
Sandeep Chakraborty (Corresponding author)
References 1. Chakraborty S: DOCLASP - Docking ligands to target proteins using spatial and electrostatic congruence extracted from a known holoenzyme and applying simple geometrical transformations [v1; ref status: awaiting peer review, http://f1000r.es/48g]. F1000Research. 2014; 3 (262). Publisher Full Text | Reference Source 2. Chakraborty S: Correlating the ability of VP24 protein from Ebola and Marburg viruses to bind human karyopherin to their immune suppression mechanism and pathogenicity using computational methods [v1; ref status: awaiting peer review, http://f1000r.es/4o3]. F1000Research. 2014; 3 (265). Publisher Full Text | Reference Source 3. Lee JE, Fusco ML, Hessell AJ, Oswald EB, et al.: Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008; 454 (7201): 177-182 PubMed Abstract | Free Full Text | Publisher Full Text | Reference Source
Competing Interests:No competing interests were disclosed.Close
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
How to fix it
Save downloaded CSV file
Open spreadsheet program (e.g. Excel)
Click the ‘Data’ tab at the top
Click the ‘From text’ icon (top left)
Browse for downloaded CSV file, click ‘Import’
Ensure ‘Delimited’ radio button is selected, click ‘Next’
Check one of the appropriate delimiter checkboxes (you can visualize the formatting by looking at the data preview below these options)
Chakraborty S, Rao BJ, Asgeirsson B and Dandekar AM. Dataset 1 in: Characterizing alpha helical properties of Ebola viral proteins as potential targets for inhibition of alpha-helix mediated protein-protein interactions. F1000Research 2014, 3:251 (https://doi.org/10.5256/f1000research.5573.d37453)
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)